
Abivax addresses safety, first Treg approval, and more
Weekly Readout #12: Week ending July 3, 2026

Disclaimer: This newsletter is for educational and informational purposes only and does not constitute medical, investment, financial, or legal advice, nor does it establish a provider-patient or advisory relationship. Content is based on publicly available topline data and includes forward-looking statements regarding investigational therapeutic candidates; their ultimate safety, efficacy, and clinical outcomes are unpredictable and remain subject to final regulatory determination. While we strive for accuracy, all information is provided “as is” without guarantees of completeness or timeliness. The author holds no direct equity positions in the specific companies mentioned in this issue nor receives third-party compensation for this coverage. Consult qualified professionals before making any medical or financial decisions. Please find a complete version of our disclaimers at the bottom of this article and on our About page.

Press ⬇️ to go down to a section and ⬆️ to go back up to the Table of Contents
Table of Contents
This week, we discuss:
China Biotech
Acquisitions ⬇️
United Therapeutics to Acquire Thymmune, regenerative medicine titans join forces to grow a thymus
Ipsen to Acquire Memo, neutralizing the viral ghosts that haunt kidney transplant patients
Ipsen to Acquire Kartos, unleashing apoptotic p53 on myelofibrosis
Zymeworks to Acquire Theravance, M&A alchemy with a COPD sweetener
Approvals
Clinical Trial Data
Abivax - Obefazimod (miR-124 upregulator) / Phase 3 (UC) ⬇️
RevMed - Zoldonrasib (KRAS-G12D inhibitor) / Phase 1 (G12D mutated mPDAC) ⬇️
AbbVie - Epkinly (CD3 x CD20 mAb) / Phase 3 (DLBCL) ⬇️
AstraZeneca - Efzimfotase alfa (TNSALP ERT) / Phase 3 (hypophosphatasia ages 2-11) ⬇️
BeOne - Brukinsa (BTK inhibitor) / Phase 3 (1L MCL) ⬇️
Roche - Divarasib (reversible KRAS G12C inhibitor) / Phase 3 (2L NSCLC) ⬇️
Otsuka - Voyxact (anti-APRIL mAb) / Phase 3 (IgAN) ⬇️
Sanofi - Nexviazyme (M6P-GAA ERT) / Phase 3 (infant Pompe) ⬇️
China Biotech
Big Pharma Partnerships Draw Scrutiny
On June 30, 2026, Endpoints reported that the U.S. House Select Committee on China has opened a national security investigation into several major American pharmaceutical companies regarding their clinical trial operations in China. This specifically includes Bristol Myers Squibb, along with Merck, AbbVie, Pfizer, and Eli Lilly. Led by Chairman John Moolenaar (R-Mich.), the committee is investigating U.S. drugmakers for sponsoring clinical trials at Chinese military hospitals and medical centers, as well as in the Xinjiang region. According to the committee’s letters, lawmakers expressed concern that these trials could potentially present ethical risks and data security vulnerabilities that could inadvertently fuel China’s military biotechnology research. Public data from ClinicalTrials.gov revealed that Bristol Myers Squibb has sponsored at least 17 trials involving PRC military medical centers and at least eight trials in Xinjiang, where they allege is “where the Chinese Communist Party (CCP) is conducting a [redacted] of Uyghur Muslims and other minorities.” The trials involved drugs for conditions like multiple sclerosis and lupus. While companies like Bristol Myers, Merck, and AbbVie are facing heavy scrutiny, the committee commended Pfizer’s CEO, Albert Bourla, following indications that Pfizer plans to stop sponsoring trials in Xinjiang and at PRC military hospitals. The committee explicitly stated they are not accusing the drugmakers of illegal activity, but rather pointing out severe ethical and security blind spots. The panel has demanded that the companies hand over details on their due diligence processes, intellectual property protections, and trial data guidelines by July 17. This probe comes amid an escalating legislative push by the U.S. Federal Government to reduce U.S. biotech reliance on China. Lawmakers have previously introduced measures to block the FDA from accepting clinical data generated at trial sites in China to incentivize companies to bring drug development back to the U.S.
Sources: Endpoints article
FDA’s Push to Re-shore Drug Manufacturing Achieves a Major Milestone
This news doesn’t refer to China specifically, but more broadly relates to re-shoring of drug manufacturing in an effort to reduce supply chain reliance on foreign countries for essential healthcare needs. The PreCheck Pilot Program was launched on February 1, 2026, directly responding to Executive Order 14293 signed by President Trump on May 5, 2025 which aimed to provide regulatory relief and promote the domestic production of critical medicines. On June 29, 2026, the U.S. Food and Drug Administration (FDA) announced the selection of seven pharmaceutical and biotechnology companies to participate in the inaugural cohort of its FDA PreCheck Pilot Program. The seven chosen companies represent a mix of massive multinationals and emerging biotechs spanning various medical fields:
Amneal Pharmaceutical (Long Island, NY): Will manufacture small molecule sterile liquid products for pain management, respiratory, and ophthalmic diseases.
Cellares Corp. (Bridgewater, NJ): Will specialize in cell-based gene therapy products for oncology and hematology.
Eli Lilly and Company (Lebanon, IN): Will manufacture active pharmaceutical ingredients (APIs) to support its existing and future medicines, including metabolic/GLP-1 drugs.
FUJIFILM Biotechnologies (Holly Springs, NC): Will support commercial-scale cell culture biomanufacturing.
Kriya Therapeutics, Inc. (Durham, NC): Will manufacture AAV-based gene therapy products to address chronic conditions.
Kyowa Kirin, Inc. (Sanford, NC): Will manufacture biotechnology drug substance targeting rare diseases.
Regeneron Pharmaceuticals, Inc. (Saratoga Springs, NY): Will manufacture biotechnology drug substances, sterile injectables, and novel protein therapeutics.
As congressional panels simultaneously probe U.S. Pharma reliance on Chinese clinical and manufacturing sites, this pilot serves as the “carrot” to Congress’s “stick.” The FDA’s selection of these partners is a concrete step toward building a self-sustaining, highly advanced domestic drug manufacturing ecosystem designed to foster national resilience.
Sources: FDA press release, Executive Order 14293

Acquisitions

Approvals
Orca Bio - Tregzi (allo HSCT) / Approved (cGVHD)
On June 30, 2026, the U.S. Food and Drug Administration (FDA) made a landmark decision by approving Tregzi, a precision-engineered cellular immunotherapy developed by Orca Bio. This milestone marks the first and only approved regulatory T-cell (Treg) therapy designed to improve survival free from chronic graft-versus-host disease (cGVHD) in adults undergoing allogeneic hematopoietic stem cell transplantation (allo-HSCT). Unlike a traditional stem cell transplant where a mixed bag of donor cells is infused directly into the patient, Tregzi is a personalized treatment formulated from a matched donor’s blood. It isolates and sequentially delivers three distinct components:
Hematopoietic stem and progenitor cells (HSPCs) to rebuild and reconstitute the patient’s blood-forming system.
Highly purified Regulatory T cells (Tregs) tasked with maintaining immune tolerance and actively suppressing GVHD.
Conventional T cells (Tcons), which are infused slightly later to eliminate any remaining cancer cells (the graft-versus-leukemia effect) and protect against infections.
The approval was driven by statistically significant data from the Phase 3 PRECISION-T clinical trial, which randomized 187 adult patients to receive either Tregzi or a standard hematopoietic stem cell transplantation. Approximately 78% of patients treated with Tregzi were alive and free from moderate-to-severe cGVHD after 1 year, compared to just 38.4% in the standard transplant control group (see graph below). The cumulative incidence of moderate-to-severe cGVHD was slashed to 12.6% in the Tregzi arm from 44% in the control group. Furthermore, the safety profile of Tregzi was strong and consistent with the toxicities typically expected during a stem cell reset (such as mouth sores and standard infections), but with lower non-relapse mortality and fewer serious infections than the traditional protocol. Fully capitalized for its upcoming launch, Orca Bio has already established reimbursement pathways across commercial and government programs to help eligible patients access this practice-changing therapy.
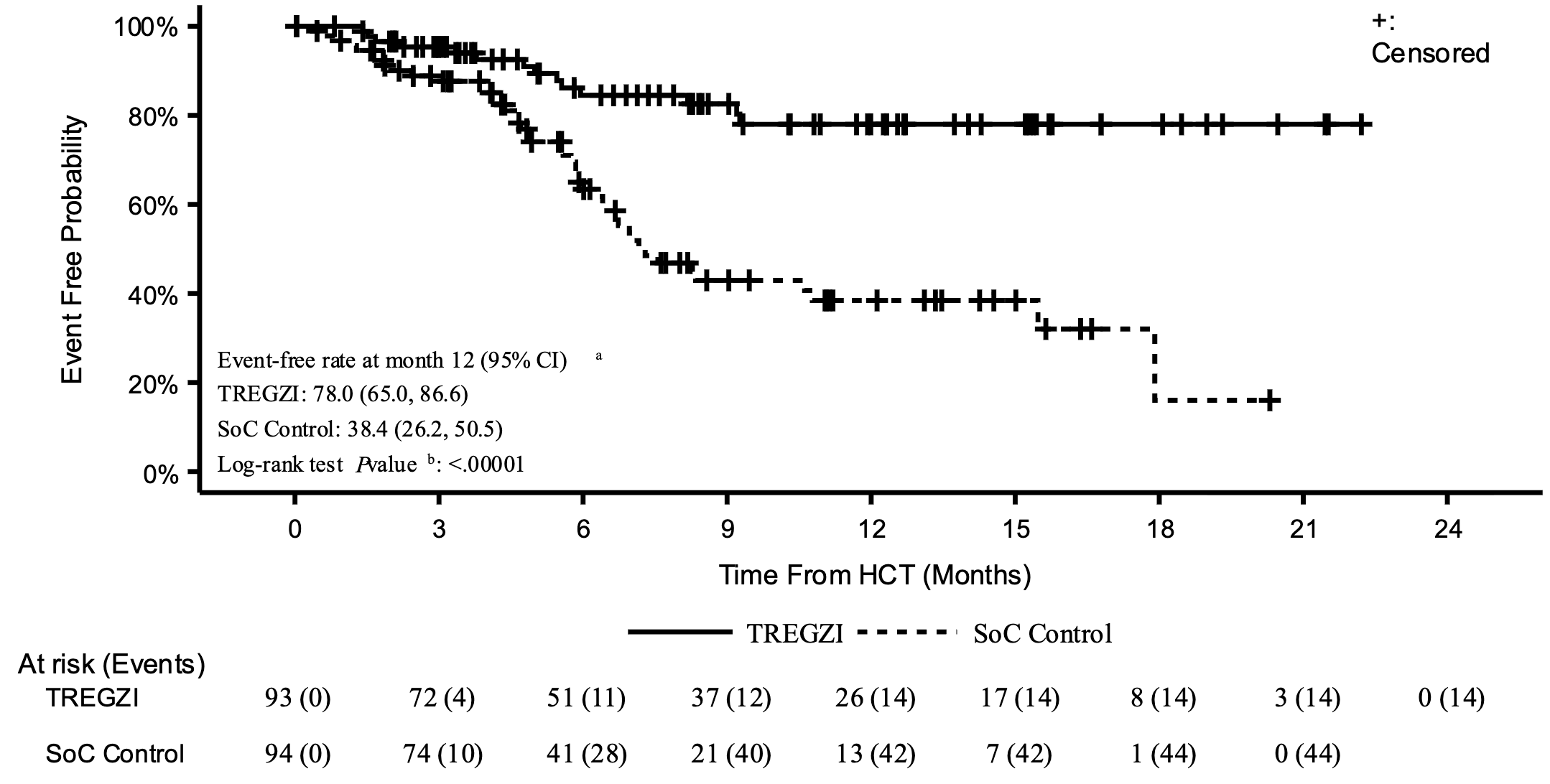
Sources: Orca Bio press release, Tregzi label
Viridian - Lumvoa (IGF-1R inhibitor) / Approval (TED)
On June 26, 2026, the U.S. Food and Drug Administration (FDA) approved Viridian Therapeutics’ Lumvoa (veligrotug-vvze) for the treatment of Thyroid Eye Disease (TED). This landmark regulatory milestone represents a paradigm shift for patients and clinicians alike. Lumvoa is the first direct competitor to Amgen’s blockbuster drug Tepezza, which was previously the only FDA-approved therapy for the condition since 2020. TED is a rare, debilitating autoimmune disorder that causes tissues behind the eyes to inflame and expand. This results in painful, disfiguring eye bulging (proptosis) and double vision (diplopia), profoundly impacting a patient’s quality of life. While both Lumvoa and Tepezza target the insulin-like growth factor-1 receptor (IGF-1R) pathway, Lumvoa enters the market with several distinct, patient-centric advantages:
The First Broad-Spectrum Label: Lumvoa is the first approved TED treatment to explicitly include data for both active and chronic stages of the disease directly on its label, giving physicians clear guidance to treat patients regardless of how long they have suffered from the condition.
A Faster, Shorter Dosing Schedule: Lumvoa provides a more efficient treatment course than historical options, requiring only five intravenous (IV) infusions administered over a 12-week period (one infusion every three weeks), compared to Tepezza’s standard eight-infusion regimen.
Superior Double Vision Resolution: Clinical results from its pivotal Phase 3 THRIVE and THRIVE-2 trials demonstrated rapid, significant reductions in eye bulging as early as three weeks, alongside unprecedented rates of complete double vision resolution.
Lumvoa’s approval was supported by two pivotal, global Phase 3 clinical trials: THRIVE and THRIVE-2. Both trials met their primary and all secondary endpoints at the 15-week primary analysis mark following a shorter, 12-week course of 5 intravenous (IV) infusions (10 mg/kg administered every 3 weeks). In the THRIVE Trial, Lumvoa achieved a 70% proptosis responder rate (defined as a ≥ 2 mm reduction in eye bulging without worsening in the fellow eye) compared to just 5% in the placebo group (see graph below). Lumvoa demonstrated a highly significant 56% proptosis responder rate by Hertel measurements, compared to 8% for placebo (a 48% placebo-adjusted difference, p < 0.0001; see graph below). In the chronic population (THRIVE-2), 56% of Lumvoa patients achieved a significant improvement (at least a 1-grade reduction on the Gorman scale) compared to 25% for placebo. Onset was rapid, showing divergence by week 6. Crucially, 32% of Lumvoa patients achieved complete resolution of their double vision (a Gorman score of 0) compared to 14% in the placebo arm (p = 0.0152). Lumvoa is immediately available for physicians to prescribe. To streamline adoption, the launch is fully supported by ViridianCares, a comprehensive hub providing insurance verification, coverage support, and patient co-pay assistance.
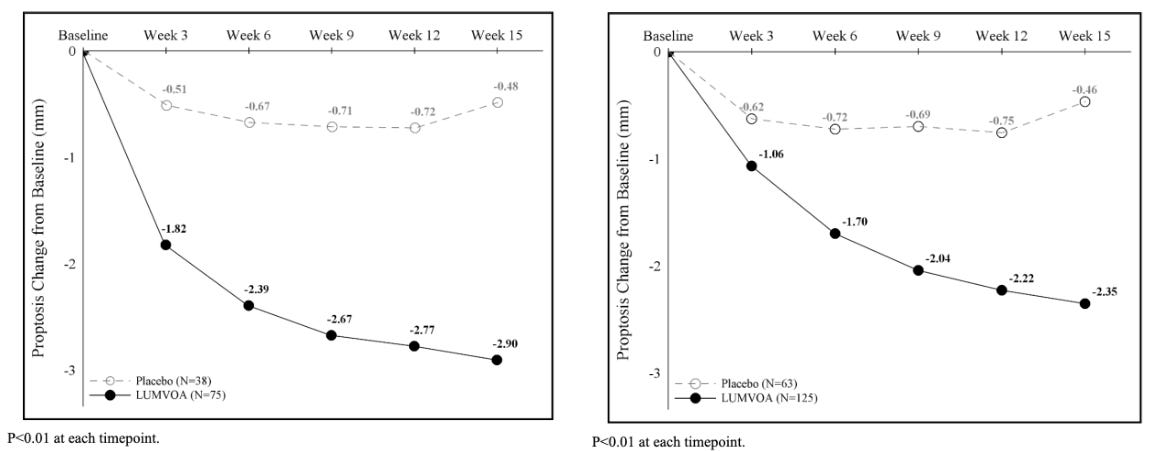
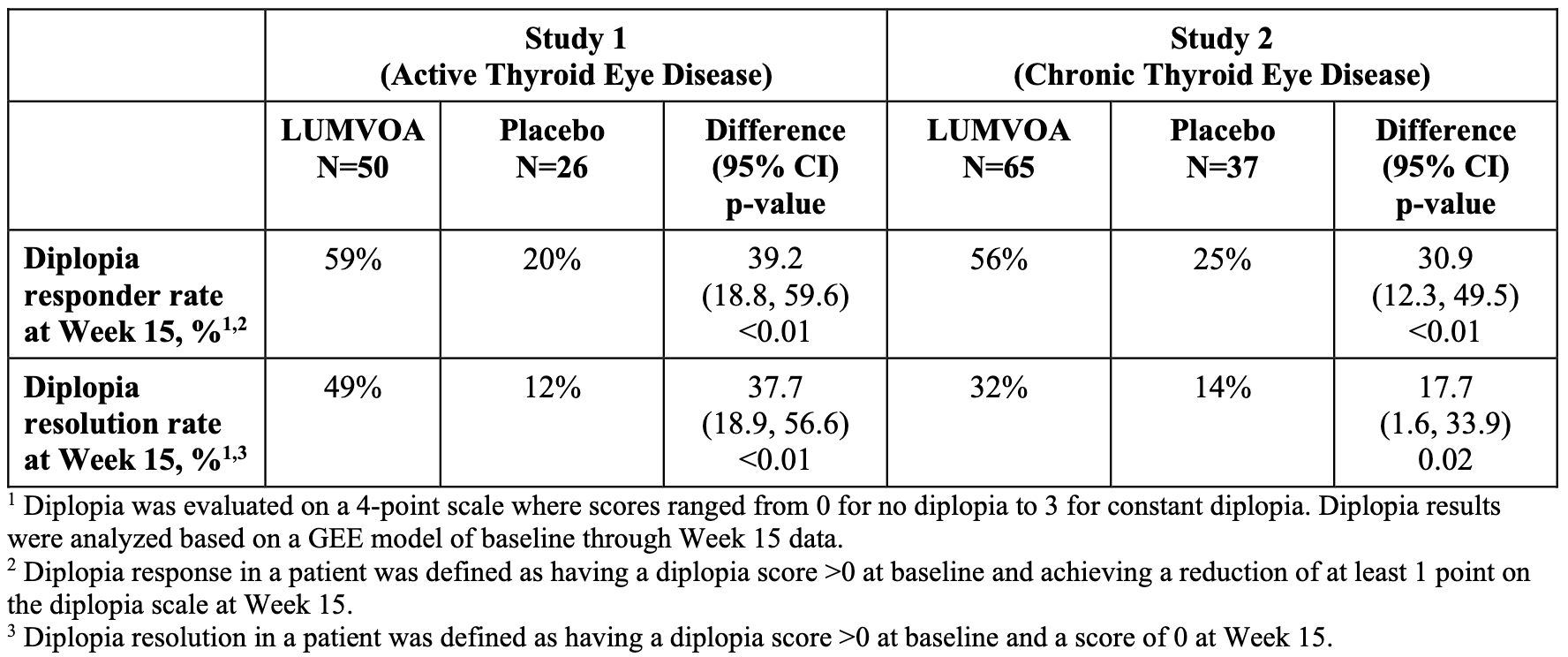
Sources: Viridian press release, Viridian slide deck, Lumvoa label
Vertex - Casgevy (BCL11A-KO HSCT) / Label Expansion (SCD/TDT age 2+)
On July 1, 2026, the U.S. Food and Drug Administration (FDA) granted a major label expansion for Vertex Pharmaceuticals and CRISPR Therapeutics’ Casgevy (exagamglogene autotemcel), extending its indication to include children as young as 2 years old. Previously restricted to patients aged 12 and older, Casgevy is now the first and only CRISPR-based gene-editing therapy approved for young pediatric patients suffering from either severe sickle cell disease (SCD) with recurrent vaso-occlusive crises (VOCs) or transfusion-dependent β-thalassemia (TDT). Both SCD and TDT stem from genetic defects in adult hemoglobin. Casgevy bypasses the mutated adult gene entirely by utilizing ex vivo CRISPR/Cas9 technology to edit a patient’s own hematopoietic stem and progenitor cells. The therapy precisely targets and knocks out the erythroid-specific enhancer region of the BCL11A gene. BCL11A acts as a genetic switch that normally shuts down the production of fetal hemoglobin (HbF) shortly after birth. By knocking out this switch, Casgevy permanently reactivates high-level HbF production. In SCD patients, the robust presence of HbF prevents red blood cells from sickling. In TDT patients, it corrects the severe hemoglobin deficiency, eliminating the root causes of both diseases.
The landmark expansion was supported by data from the ongoing Phase 3 open-label CLIMB-141 and CLIMB-151 pediatric trials. While Vertex initially sought approval for ages 5 to 11, the striking consistency of the data prompted the FDA to expand the label down to age 2:
Sickle Cell Disease Cohort (Ages 5 to <12): The FDA evaluated 11 young patients with severe SCD. Out of the 8 patients who reached the timeframe to be evaluable for efficacy, 100% (8/8) achieved complete freedom from severe vaso-occlusive crises (VOC-free status) for at least 12 consecutive months within the first two years post-infusion.
β-Thalassemia Cohort (Ages 5 to <12): In a trial of 15 pediatric patients with TDT, 88.9% (8/9) of the efficacy-evaluable children achieved complete transfusion independence for at least 12 consecutive months, boasting a median duration of 20.1 months without needing a single blood transfusion.
Extrapolation to Ages 2-4: Since trials for children aged 2 to 4 were still ongoing but showing nearly identical biological and product characteristics, the FDA utilized clinical extrapolation. Recognizing the highly predictable consistency of the gene-editing mechanism across age groups, regulators simultaneously granted the wider age clearance.
The approval was processed with remarkable speed through the FDA’s Commissioner’s National Priority Voucher (CNPV) pilot program, which shaved traditional 10-to-12-month review timelines down to just 53 days from the official filing date.
Sources: Vertex press release, updated Casgevy label
Arcutis - Zoryve (topical PDE4 inhibitor) / Label Expansion (plaque psoriasis age 2+)
In a significant advancement for pediatric dermatology, the U.S. Food and Drug Administration (FDA) has granted a major label expansion for Arcutis Biotherapeutics’ Zoryve (roflumilast) cream 0.3%. Previously indicated only for individuals aged 6 and older, Zoryve is now approved for the treatment of mild, moderate, and severe plaque psoriasis in pediatric patients as young as 2 years old. Plaque psoriasis in toddlers and young children can be exceptionally difficult to manage. The disease frequently presents in highly sensitive, thin-skin regions, such as the face, neck, and skin folds (intertriginous areas), where traditional mid-to-high-potency topical steroids are strictly restricted due to the risk of skin thinning, systemic absorption, and stretch marks. This expansion provides families and pediatricians with a much-needed, steroid-free alternative.
The regulatory clearance was supported by a comprehensive package demonstrating consistent safety and efficacy across age groups. Initial approvals in adults and older children were established by the twin, pivotal DERMIS-1 and DERMIS-2 Phase 3 trials, which demonstrated rapid plaque clearance, marked reduction in itch, and a highly favorable safety profile. To expand the label down to age 2, Arcutis conducted a specific open-label Phase 3 maximal usage pharmacokinetics (PK) study in toddlers. The data confirmed that the 0.3% dose was excellently tolerated, showed a predictable and safe systemic exposure profile identical to older demographics, and generated no new safety signals. Arcutis has ensured immediate commercial availability for this expanded age group, backed by its existing commercial access programs to minimize out-of-pocket friction for families seeking a steroid-free option for their young children.
Sources: Arcutis press release, updated Zoryve label
Clinical Trial Data
Abivax - Obefazimod (miR-124 upregulator) / Phase 3 (UC)
On June 29, 2026, Abivax reported highly anticipated topline data from Part 2 of its Phase 3 ABTECT maintenance trial for obefazimod in moderate-to-severe ulcerative colitis (UC). This readout addressed lingering market anxieties regarding a potential malignancy signal from the earlier Part 1 readout, positioning the asset for a planned regulatory submission.
Ulcerative colitis (UC) is a chronic inflammatory bowel disease characterized by continuous mucosal inflammation of the colon, driven by a dysregulated immune response against intestinal microbiota that triggers epithelial barrier breakdown and an overproduction of pro-inflammatory cytokines (TNF-α, IL-6, IL-12/23). While standard treatment escalates from anti-inflammatory 5-ASAs and corticosteroids for mild disease to biologics and oral small molecules (JAK or S1P inhibitors) for moderate-to-severe cases, obefazimod introduces a potential first-in-class, oral mechanism of action to this therapeutic landscape. By selectively binding to the cellular cap-binding complex (CBC) 80/20, obefazimod enhances RNA splicing to upregulate microRNA-124 (miR-124), which functions as a physiological brake that systematically suppresses key pro-inflammatory cytokines (IL-6, TNF-α, IL-17) and limits pathogenic Th17 cell proliferation, effectively downregulating colonic inflammation without inducing broad, systemic immunosuppression.
The Phase 3 ABTECT maintenance program evaluated daily oral obefazimod (25 mg and 50 mg) against a placebo in moderate-to-severe ulcerative colitis (UC) across two parts: a 44-week randomized responder re-randomization trial (Part 1, n=580) and an expansion study in refractory or relapsed cohorts (Part 2). Part 2 demonstrated that dose optimization with continuous 50 mg obefazimod rescued 37.2% of initial induction non-responders into clinical remission and recaptured clinical remission in approximately 45% of patients who had relapsed during Part 1. Crucially, Abivax reported a pooled analysis of 1,704 patient-years of exposure demonstrating a malignancy rate (0.56 IR/100 PY) that the company noted falls within the expected UC background range (0.30–0.70 IR/100 PY), with non-melanoma skin cancers isolated to older patients with pre-existing risk factors.
Abivax is fully capitalized and remains on track to submit its formal New Drug Application (NDA) to the U.S. FDA for obefazimod in ulcerative colitis. The company anticipates the topline data readout from its Phase 2b ENHANCE-CD induction trial in mid-2027, which is currently evaluating obefazimod’s clinical utility in Crohn’s disease with the goal of expanding its IBD franchise footprint.
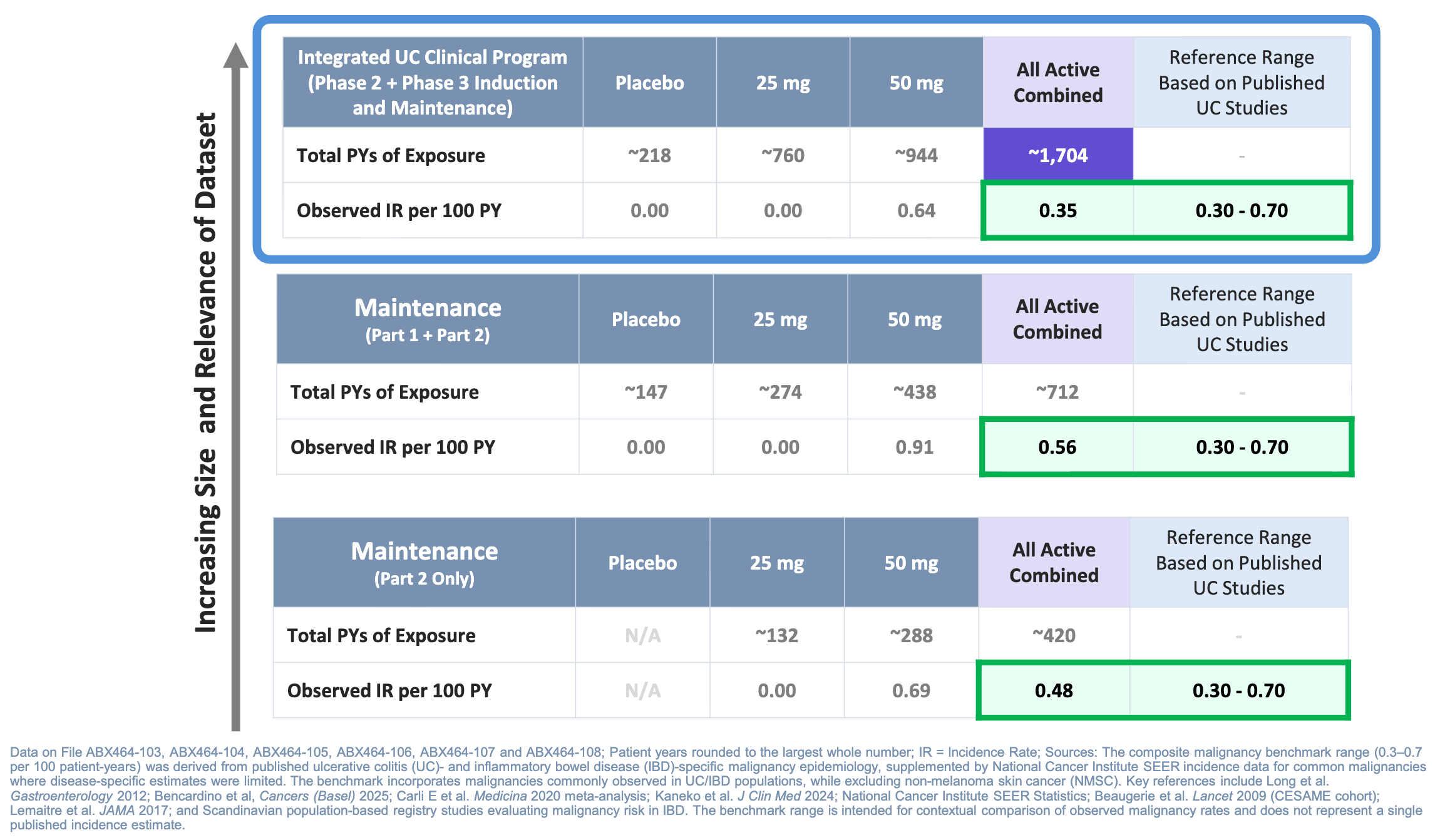
Sources: Abivax press release, Abivax slide deck
RevMed - Zoldonrasib (KRAS-G12D inhibitor) / Phase 1 (G12D mutated mPDAC)
At the 2026 European Society for Medical Oncology (ESMO) Gastrointestinal Cancers Congress, Revolution Medicines presented encouraging Phase 1/2 data for its candidate zoldonrasib (RMC-9805) in combination with its multi-RAS inhibitor daraxonrasib (RMC-6236). This readout established early signs of clinical efficacy in heavily pretreated, advanced KRAS G12D-mutant metastatic pancreatic ductal adenocarcinoma (mPDAC), a disease historically resistant to standard therapies.
KRAS G12D-mutated metastatic pancreatic ductal adenocarcinoma (mPDAC) is an aggressive malignancy where a specific missense mutation permanently locks the KRAS protein into an active, GTP-bound “ON” conformation, continuously fueling downstream proliferative pathways (MAPK/ERK, PI3K/AKT) and leaving patients with limited standard options beyond intense systemic chemotherapies like FOLFIRINOX or gemcitabine/nab-paclitaxel. Addressing this historically untargetable driver, zoldonrasib introduces a first-in-class, oral, mutant-selective mechanism that directly targets the active “ON” state of the mutated KRAS G12D protein. By utilizing a molecular glue approach, zoldonrasib binds the active KRAS G12D mutant and an abundant intracellular chaperone protein, cyclophilin A, to form a stable tri-complex. This steric block selectively modifies the mutant aspartic acid residue to turn off hyperactive MAPK signaling and prevent downstream effector interaction while minimizing disruption of wild-type RAS variants or harm to healthy tissue.
The multicenter, open-label Phase 1 RMC-9805-001 trial evaluated a daily oral combination of the mutant-selective covalent RAS(ON) G12D inhibitor zoldonrasib (1200 mg) and the pan-RAS(ON) inhibitor daraxonrasib (300 mg) in 60 patients with advanced, chemotherapy-refractory KRAS G12D-mutant metastatic pancreatic ductal adenocarcinoma (mPDAC). In the second-line setting (n=30), the combination achieved a 50% objective response rate (ORR), a 97% disease control rate (DCR), and an unprecedented median progression-free survival (mPFS) of 9.6 months, while maintaining a 47% ORR and 7.6 months mPFS in third-line or later patients (n=30). Furthermore, the combination exhibited a predictable and manageable safety profile. Treatment-emergent adverse events were primarily mild-to-moderate (Grade 1/2) on-target toxicities such as rash (90%), diarrhea (63%), nausea (57%), and stomatitis (53%). The combination also had low discontinuation rates and minimal severe toxicities. Building on this foundational late-line success, Revolution Medicine has pushed zoldonrasib into earlier settings. At the same congress, the company detailed its front-line (1L) combination strategies with chemotherapy (mFOLFIRINOX and GnP), which achieved staggering front-line ORRs up to 82%.
Revolution Medicine has initiated the global, randomized Phase 3 RASolute 305 trial. This study will evaluate zoldonrasib directly alongside investigator’s choice of standard chemotherapy backbones in treatment-naïve, first-line metastatic KRAS G12D PDAC patients, laying the groundwork for a future definitive regulatory approval.
Sources: RevMed press release
AbbVie - Epkinly (CD3 x CD20 mAb) / Phase 3 (R/R DLBCL)
On July 3, 2026, AbbVie and Genmab announced positive topline results from their Phase 3 EPCORE DLBCL-1 clinical trial evaluating Epkinly (epcoritamab-bysp) in patients with relapsed or refractory (R/R) diffuse large B-cell lymphoma (DLBCL). This successful readout provides a major clinical victory and redemption for the asset’s late-line franchise after it missed an overall survival milestone earlier in the year.
Diffuse large B-cell lymphoma (DLBCL) is an aggressive hematologic malignancy characterized by the uncontrolled clonal proliferation of malignant B-lymphocytes, driven by chromosomal translocations (MYC, BCL2, BCL6) and dysregulated signaling pathways (NF-κB) that block cellular differentiation. While frontline treatment relies on intense chemoimmunotherapy like R-CHOP or Polivy-R-CHP, and later-line options shift based on transplant eligibility to salvage chemotherapy, CAR-T, or targeted agents, Epkinly (epcoritamab-bysp) introduces a highly targeted therapeutic approach. As a first-in-class, subcutaneous CD3×CD20 T-cell engaging bispecific antibody, Epkinly simultaneously binds to CD20 on malignant B-cells and CD3 on mature T-cells. By physically bridging these cells, it bypasses traditional major histocompatibility complex (MHC) class I restrictions to trigger the direct activation of endogenous T-cells, which release cytotoxic perforins and granzymes to induce immunologically driven lysis of the lymphoma cells. Check out CAR-T, Part 2 if you are interested in learnings more about the history and cutting edge of TCEs.

The global, randomized, open-label Phase 3 EPCORE DLBCL-1 trial evaluated subcutaneous Epkinly monotherapy against standard salvage chemotherapy (primarily the R-GemOx regimen) in patients with relapsed or refractory (R/R) diffuse large B-cell lymphoma (DLBCL) who were transplant-ineligible and had failed at least two prior systemic lines. Topline data demonstrated that Epkinly met its primary endpoint by remarkably reducing the risk of disease progression or death by 60% (HR=0.40) compared to standard chemotherapy, while maintaining a predictable safety profile with low-grade, manageable cytokine release syndrome (CRS) confined to the initial step-up dosing weeks. This decisive victory rehabilitates the Epkinly franchise after an earlier 2026 second-line clinical trial setback.
AbbVie and Genmab have announced they will immediately engage with global regulatory authorities (including the U.S. FDA and European Medicines Agency) to submit these data to expand Epkinly‘s label from its current accelerated status into a full, definitive regulatory approval for late-line DLBCL. The next major inflection point for the franchise will occur later in 2026, when AbbVie expects to read out pivotal Phase 3 data evaluating Epkinly moved up into first-line (1L) newly diagnosed DLBCL in combination with standard R-CHOP chemotherapy.
Sources: AbbVie press release, Genmab press release
AstraZeneca - Efzimfotase alfa (TNSALP ERT) / Phase 3 (hypophosphatasia ages 2-11)
On July 2, 2026, AstraZeneca announced positive topline results from its global, randomized Phase 3 clinical trial evaluating efzimfotase alfa (ALXN1850) in pediatric patients aged 2 to 11 with hypophosphatasia (HPP). This pediatric readout establishes a distinct clinical data package for the asset, which previously did not meet its primary endpoints in a separate Phase 3 trial evaluating adolescents and adults.
Hypophosphatasia (HPP) is a rare, inherited metabolic disorder caused by loss-of-function mutations in the ALPL gene that result in a deficiency of tissue-nonspecific alkaline phosphatase (TNSALP), leading to an extracellular accumulation of inorganic pyrophosphate (iPP), a potent biochemical inhibitor of hydroxyapatite crystal formation that causes rickets, progressive skeletal deformities, and muscle weakness. While standard care focuses on supportive management or the established enzyme replacement therapy (ERT) Strensiq (asfotase alfa) to restore bone mineralization, efzimfotase alfa (ALXN1850) introduces a next-generation, bone-targeted TNSALP enzyme replacement therapy to addressing this enzyme deficiency. Engineered as a recombinant soluble TNSALP fusion protein modified with a negatively charged, deca-aspartate peptide tail, efzimfotase alfa selectively binds with high affinity to hydroxyapatite crystals within the mineralizing bone matrix. Once localized, it directly cleaves accumulated iPP into inorganic phosphate, systematically removing the biochemical barrier to healthy bone architecture and mineralization.
The global, randomized, double-blind Phase 3 trial evaluated the efficacy and safety of subcutaneous efzimfotase alfa against a placebo over a 25-week primary evaluation period in 64 pediatric patients (aged 2 to 11) with pediatric-onset hypophosphatasia (HPP). Topline results demonstrated that the trial successfully met its primary endpoint, with the efzimfotase alfa cohort achieving a statistically significant mean Radiographic Global Impression of Change (RGI-C) score improvement of +1.67 points compared to 0.00 points in the placebo arm (P=0.0003), driving substantial radiographic healing of active rickets and skeletal lesions alongside robust improvements in growth velocity and motor function. The next-generation enzyme replacement therapy was well-tolerated, with mild-to-moderate injection-site reactions representing the most frequent adverse events and no severe systemic toxicities reported. This successful pediatric readout provides critical continuity for AstraZeneca’s Alexion rare disease franchise as a pipeline successor to Strensiq, demonstrating a highly effective targeted therapeutic window during early childhood development when bone turnover is highest, despite the asset’s previous clinical failures in less dynamic adolescent and adult populations.
Armed with a highly significant pediatric dataset, AstraZeneca intends to discuss these results with global regulatory bodies, including the U.S. FDA and the European Medicines Agency (EMA), to seek marketing authorization specifically for pediatric HPP patients. The trial is transitioning into its open-label extension phase to monitor the long-term durability of bone mineralization, growth parameters, and antibody safety profiles out to two years post-infusion.
Sources: AstraZeneca press release
BeOne - Brukinsa (BTK inhibitor) / Phase 3 (1L MCL)
On July 3, 2026, BeOne announced positive topline results from its global Phase 3 clinical trial evaluating Brukinsa (zanubrutinib) in treatment-naïve patients with mantle cell lymphoma (MCL). This successful frontline readout marks a major therapeutic milestone, positioned to disrupt the traditional chemoimmunotherapy paradigm for newly diagnosed patients.
Mantle cell lymphoma (MCL) is an aggressive B-cell malignancy driven by the hallmark t(11;14) translocation that causes constitutive overexpression of cyclin D1, forcing unchecked cell cycle progression, which is further sustained by hyperactive B-cell receptor (BCR) signaling mediated by Bruton’s tyrosine kinase (BTK). The frontline standard of care is strictly stratified by patient fitness, relying on intensive cytarabine-based chemoimmunotherapy and autologous stem cell transplantation (ASCT) for younger patients, and regimens like bendamustine plus rituximab (BR) for transplant-ineligible individuals. As a next-generation, oral small-molecule BTK inhibitor, Brukinsa forms an irreversible, covalent bond within the ATP-binding pocket of the BTK enzyme to profoundly disrupt downstream proliferative networks (MAPK, NF-κB). It was specifically engineered to maximize target occupancy while minimizing off-target inhibition of structurally similar kinases (such as EGFR, TEC, and ITK), thereby turning off oncogenic survival signals while successfully reducing common class-related toxicities.
The global, randomized, open-label Phase 3 trial evaluated oral Brukinsa (zanubrutinib) monotherapy as a chemotherapy-free alternative against standard investigator-chosen chemoimmunotherapy (such as bendamustine plus rituximab) in previously untreated, transplant-ineligible adult patients with newly diagnosed mantle cell lymphoma (MCL). Topline data demonstrated that frontline Brukinsa significantly met its primary endpoint, cutting the risk of disease progression or death by 43% (HR=0.57) while showcasing a favorable selectivity profile with low rates of cardiac adverse events and significantly lower rates of severe myelosuppression and febrile neutropenia compared to the chemotherapy arm. Secondary endpoints, including objective response rate and a positive trend in overall survival, also favored Brukinsa.
BeOne plans to present the detailed, granular survival and response data at an upcoming major medical congress and will initiate discussions with global regulatory authorities (including the U.S. FDA and EMA) to file supplemental regulatory submissions for expanded frontline approval. Moving forward, next steps will focus on exploring Brukinsa-led combinations in earlier lines of therapy, including ongoing trials evaluating its synergy with BCL-2 inhibitors (such as venetoclax) to achieve deeper, fixed-duration molecular remissions.
Sources: BeOne press release
Descriptive data releases without numerical data
Roche - Divarasib (reversible KRAS G12C inhibitor) / Phase 3 (2L NSCLC): The Phase 3 Krascendo-1 trial evaluating Roche’s divarasib, a reversible KRAS G12C inhibitor, achieved a statistically significant and clinically meaningful success in the second-line relapsed or refractory non-small cell lung cancer (2L NSCLC) setting. The trial demonstrated that divarasib delivered robust improvements in both overall survival (OS) and progression-free survival (PFS) when compared directly against the investigator’s choice of standard covalent KRAS G12C inhibitors, specifically Amgen’s Lumakras (sotorasib) or Bristol Myers Squibb’s Krazati (adagrasib). While detailed data from this readout are slated for presentation at an upcoming medical conference, Roche is already advancing the asset’s clinical pipeline, with the Krascendo-2 trial actively ongoing to evaluate divarasib’s therapeutic potential in the frontline (1L NSCLC) setting.
Otsuka - Voyxact (anti-APRIL mAb) / Phase 3 (IgAN): Otsuka announced positive topline results from its Phase 3 trial evaluating Voyxact (sibeprenlimab-szsi), a first-in-class anti-APRIL monoclonal antibody, in patients with primary IgA nephropathy (IgAN). The trial successfully met its key long-term objectives, demonstrating that Voyxact treatment led to a statistically significant and clinically meaningful stabilization of kidney function after two years of follow-up compared to placebo. These critical 24-month data, slated for full presentation at an upcoming medical conference, establish the durable clinical efficacy of the therapy and serve as the foundational evidence required to secure full regulatory approval.
Sanofi - Nexviazyme (M6P-GAA ERT) / Phase 3 (infant Pompe): The Phase 3 trial evaluating Sanofi’s Nexviazyme (avalglucosidase alfa), a next-generation, mannose-6-phosphate-enriched receptor-targeted acid alpha-glucosidase (M6P-GAA) enzyme replacement therapy, achieved a clinical success in treatment-naïve, infant-onset Pompe disease. The study successfully met its primary efficacy endpoint, which measured the percentage of treatment-naïve infants aged 6 months and younger who were alive and free from the need for invasive ventilation at 52 weeks. Although specific data metrics and numbers were not disclosed in the initial announcement, Sanofi is scheduled to present the comprehensive data from this readout on July 8 at the International Congress on Neuromuscular Diseases.
Love biotech? Check out Biotech Readout’s full content library, or navigate directly to a segment that interests you:
Frontiers in Medicine: Exploring the frontiers of our understanding and treatments for disease.
Medical History: Recovering forgotten relics in the history of medicine.
Acquisitions: Exploring the innovation behind acquired companies.
Weekly Readout: A digest of new clinical data from the past week.

To contact us, please send us an email at biotechreadout@gmail.com
Disclaimers
Investigational Status Disclaimer
The therapeutic candidates discussed in this newsletter are currently in clinical development and have not been approved for commercial sale by the U.S. Food and Drug Administration (FDA), the European Medicines Agency (EMA), or other global regulatory authorities. Their safety and efficacy have not been established. References to pipeline products and ongoing clinical trials involve significant risks and uncertainties. Statements regarding the potential safety, potency, or efficacy of investigational drugs reflect current hypotheses and are not a guarantee of future performance or regulatory clearance. The outcome of clinical trials is inherently unpredictable, and clinical results from earlier stages may not be predictive of results in later, larger-scale trials.
No Medical Advice Disclaimer
This newsletter is for informational and educational purposes only. The content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or another qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read in this publication.
No Patient-Provider Relationship Disclaimer
The information provided in this newsletter is for educational and analytical purposes only. Receipt of this information, or any interaction with this content, does not create a physician-patient, pharmacist-patient, or any other professional-provider relationship between you and the authors or publishers. This newsletter should not be used as a substitute for a personal consultation with a qualified healthcare professional.
Forward-Looking Statements Disclaimer
This newsletter contains “forward-looking statements” regarding future events, including clinical trial timing, regulatory milestones, and projected market performance. These statements are based on current expectations and assumptions that are subject to significant risks and uncertainties. Actual results may differ materially from those expressed or implied. We undertake no obligation to update these statements as a result of new information or future developments.
Third-Party Links & Content Disclaimer
This newsletter contains links to third-party websites, including clinical trial registries and corporate presentations. Biotech Readout does not endorse, guarantee, or assume responsibility for the accuracy or reliability of any information offered by third-party providers.
Errors and Omissions Disclaimer
While we strive for technical accuracy, the information in this newsletter is provided on an “as is” basis with no guarantees of completeness, accuracy, or timeliness. Biotech Readout assumes no liability for any errors or omissions in the content of this publication.
Non-Endorsement Disclaimer
Any reference to specific commercial products, processes, or services by trade name, trademark, or manufacturer does not constitute or imply an endorsement or recommendation by the author. All trademarks are the property of their respective owners.
No Investment Advice Disclaimer
This newsletter is for informational purposes only and does not constitute financial, investment, or legal advice. The author is not a registered investment advisor. You should consult with a professional financial advisor before making any investment decisions. The biotechnology sector is highly volatile; past performance is not indicative of future results.
Conflict of Interest Disclaimer
The author of this newsletter maintains a position of independence. At the time of publication, the author holds no direct financial interest, equity, or options in any of the companies mentioned in this report. No compensation has been received from any third party to feature or analyze specific therapeutic candidates or corporate entities.