
China Biotech: Feast or Famine?
Examining biotech's newest innovation engine

Disclaimer: This newsletter is for educational and informational purposes only and does not constitute medical, investment, or financial advice, nor does it establish a provider-patient relationship. Content may include forward-looking statements and discussions of investigational therapeutic candidates that are not FDA/EMA approved; their safety and efficacy remain unestablished and clinical outcomes are unpredictable. While we strive for accuracy, all information is provided as is without guarantees. This newsletter is independent, and the author holds no financial positions in the companies mentioned nor receives third-party compensation for this coverage. Please find a complete version of our disclaimers at the bottom of this article and on our About page.

Introduction
A spirited debate has emerged in late May 2026 regarding the structural impact of China Biotech on the global healthcare ecosystem. The debate centers on the competing interests of those who are generally enthusiastic about China Biotech versus those who are generally cautious of China Biotech.

My initial idea for this article was simply to summarize both sides of the debate, but my research has motivated me to go a bit further. Here, I aim to re-frame the discussion away from “should we or shouldn’t we,” and toward an examination of the potential feasts & famines that could emerge from U.S. Biotech’s deeper engagement with China Biotech. This research is organized into four lenses for looking at China Biotech:
The Good (accelerated innovation)
The OK (capturing the “me better” arbitrage)
The Bad (pharma cuts out the middleman)
The Ugly (obsolescence of U.S. biotech)

Before we dive into China Biotech, it is imperative that we recognize the most powerful driving force behind the need for relentless biotech innovation (aside from benefitting patients, of course): the patent cliff.
Note to readers: I cover new China Biotech news every week in my Weekly Readout segment. Lookout for the blue pill, and subscribe for free to stay up to speed.

Overcoming the Crushing Weight of Patent Cliffs
In the pharmaceutical industry, a patent cliff refers to the sharp, sudden drop in revenue a pharmaceutical company experiences when the patent protection on its highly profitable blockbuster drugs (drugs generating >$1 billion in annual sales) expires. Once this market exclusivity lapses, cheaper generic or biosimilar alternatives can enter the market. This exposes the original drug maker to intense competition and breaks its monopolistic pricing power. Unlike a slow transition, a patent cliff can trigger a 30% to 90% drop in brand revenue within the very first year of generic competition. Multi-billion-dollar revenue streams shift away from the innovator firm and directly benefit generic manufacturers, insurance payers, and patients through drastically lower prices.
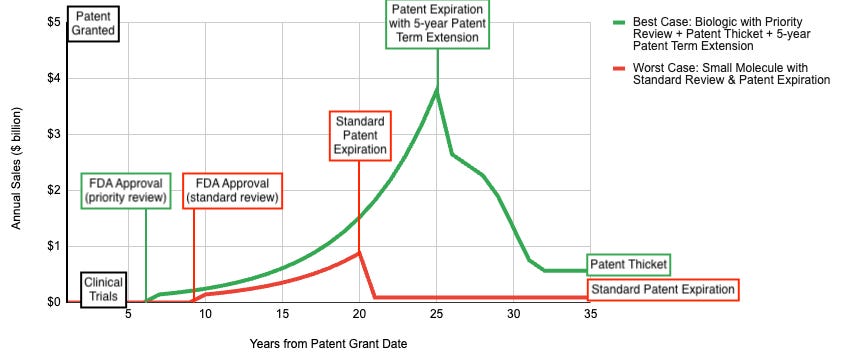
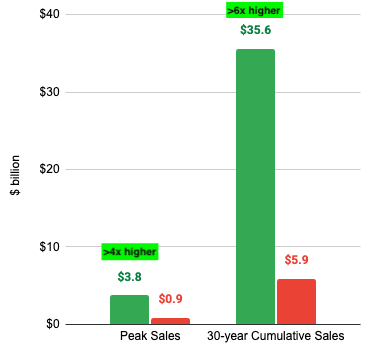
Before the mid-1980s, the concept of a patent cliff didn’t exist. When a drug’s patent expired, generic competitors had to repeat the entire, extraordinarily expensive clinical trial process from scratch to prove safety and efficacy to the Food & Drug Administration (FDA). Since the financial barrier to entry was so high, very few generic companies bothered. Even after a patent expired, branded pharmaceutical companies retained an effective monopoly for years because copying the drug was too hard.
The modern patent cliff was created by the Hatch-Waxman Act of 1984 (also called “The Drug Price Competition and Patent Term Restoration Act”). This act created the Abbreviated New Drug Application (ANDA), a submission to the FDA for the review and potential approval of a generic drug product, demonstrating that the proposed drug is equivalent in safety, efficacy, and quality to an existing licensed medication. This allowed generic manufacturers to skip clinical trials entirely. They only had to prove bioequivalence, evidence that the generic pill dissolves and delivers the exact same active ingredient to the bloodstream at the same rate as the branded version. To incentivize generic firms to challenge patents, the law granted the first generic company to successfully file an application 180 days of market exclusivity, during which the price of their drug could hover just slightly below the branded price. The ensuing decade saw a number of localized sales shocks due to patent cliffs:
The Prozac Shock (2001): Eli Lilly’s blockbuster antidepressant Prozac lost patent protection in August 2001. Driven by Hatch-Waxman efficiencies and rapid pharmacy substitution, generic competitors captured over 70% of Prozac’s market share within just two months.
The Zocor Shock (2006): Merck’s massive cholesterol drug Zocor faced a similar immediate obliteration when its patent expired, immediately transferring billions in revenue to generic manufacturers like Teva.
This was just a preview of what was to come. Following the massive success of early statins and antidepressants in the 1990s, the pharmaceutical industry pivoted to the Blockbuster Model: discover small-molecule chemical pills aimed at massive, chronic patient populations (hypercholesterolemia, hypertension, gastrointestinal disease, and depression). Since these drugs were discovered, approved, and launched during the same mid-to-late-1990s boom, their 20-year patent clocks were synchronized to run out between late 2011 and late 2013, setting off The Great Patent Cliff of 2011-2013.
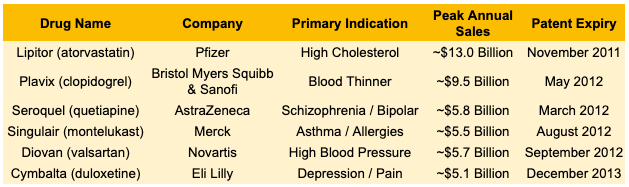
The most dramatic case study was Lipitor. As the best-selling drug in medical history up to that point, it accounted for over 20% of Pfizer’s total corporate revenue. When its patent expired on November 30, 2011, the drop was vertical. Within 180 days, generic alternatives captured the vast majority of the volume, and Lipitor‘s U.S. revenues plummeted by over 80%.
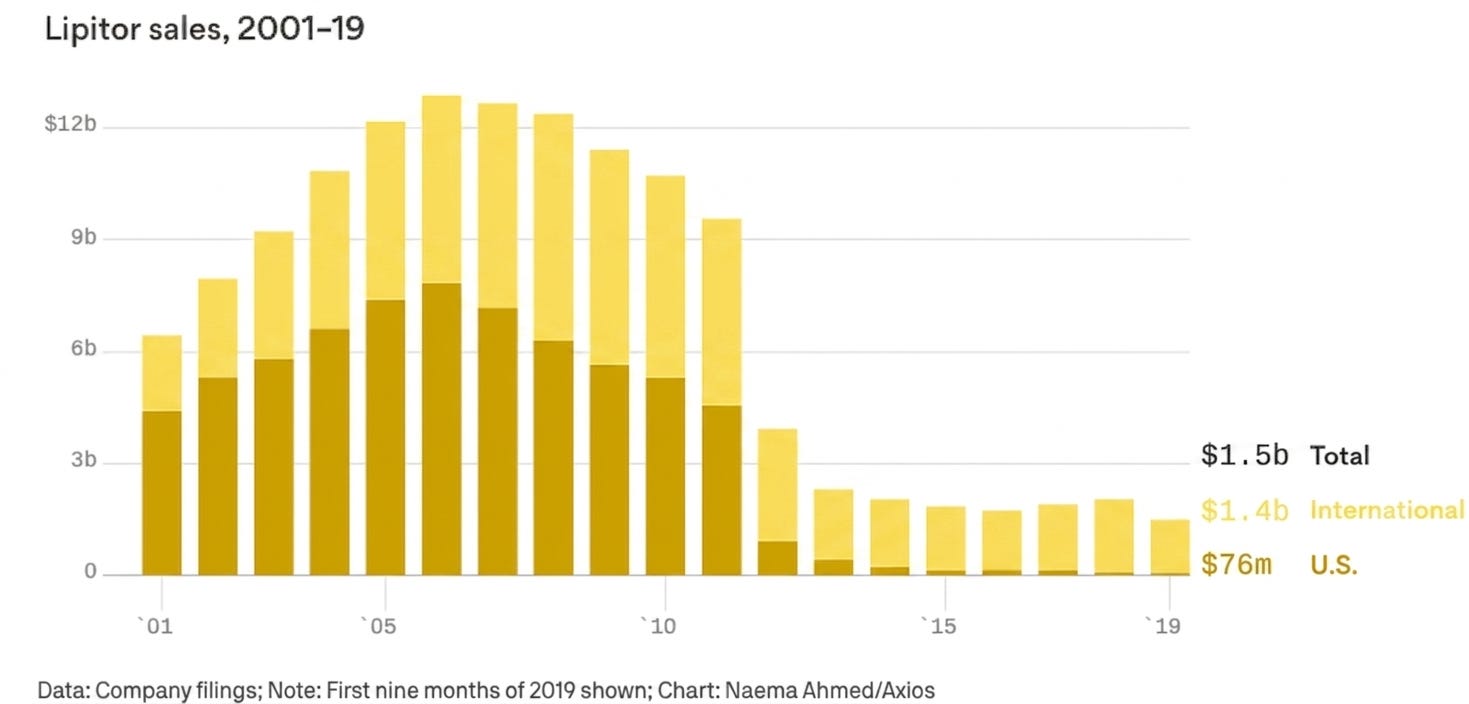
Lipitor‘s patent cliff was made significantly more brutal because of an operational shift in the U.S. healthcare system: the maturation of Managed Care and Pharmacy Benefit Managers (PBMs). Before 2011, a patient might actively ask their doctor to keep them on a brand-name drug out of habit. By 2012, automated formulary protocols within the PBM ecosystem had largely institutionalized generic substitution at the point of sale. The moment the patent expired, electronic systems could automatically prompt pharmacists to substitute the generic. If a patient insisted on the branded version, the insurer could refuse to pay for it or hit the patient with a massive, prohibitive co-pay. Faced with what many industry analysts at the time characterized as an ‘earnings black hole’, pharmaceutical CEOs had to dramatically reinvigorate their businesses to ensure their longevity. The Great Patent Cliff of 2011-2013 triggered three major industry shifts: an M&A wave, downsizing, and a pivot to biologics.
Companies realized they could not innovate their way out of the hole in a 24-month timeframe, so they bought revenue and cut duplicated corporate overhead to preserve margins. This era catalyzed massive consolidation:
Pfizer bought Wyeth for $68 billion to diversify into biologics and vaccines (acquiring Prevnar).
Merck bought Schering-Plough for $41 billion to secure late-stage pipeline assets and international revenue.
Roche bought the remaining shares of Genentech for $47 billion to fully capture its impressive oncology pipeline.
In a bid to optimize earnings per share (EPS), pharmaceutical companies realigned their internal early-stage discovery budgets. This shift frequently involved consolidating legacy primary-care research footprints and increasingly outsourcing early-stage risk to academic spinouts and small biotech startups. The most profound historical legacy of The Great Patent Cliff of 2011-2013 was where the industry chose to reinvest its capital:
Pharma decreased its dependence on small-molecule chemical pills that could be easily and cheaply copied by a generic manufacturer. Instead, they prioritized biologics like antibodies because they were vastly more difficult to copy, requiring an entirely different regulatory pathway called biosimilars, which do not suffer the same rapid 90% revenue destruction upon patent expiry.
They also moved away from primary-care mass markets (cardiorenal and neuropsych) toward specialized medicine (oncology and orphan disease), where pricing power was insulated and clinical trials were faster.
Today, pharmaceutical companies have been known to deploy a handful of strategies to navigate impending cliffs:
Patent Thickets (delaying competition): Instead of protecting a medicine with just one or two core patents on the active molecule, companies surround a blockbuster with dozens, sometimes hundreds, of secondary patents covering manufacturing methods, formulations, or specific dosages. AbbVie famously filed over 130 patents protecting Humira, creating a barrier that delayed biosimilar entry in the U.S. for years. Generic competitors must challenge each patent individually in court, which is prohibitively expensive and time-consuming, effectively forcing them to settle for delayed launch dates.
Product Hopping (internal innovation): This involves unveiling a slightly modified, next-generation version of the drug and switching patients to it before the original version goes generic. These can include extended release/long acting version that enable similar safety/efficacy but can be taken less frequently, combination pills/shots that serve as a one-stop-shop for patients who are taking multiple medications for the same disease, or more convenient routes of administration (making an in-hospital IV an at-home autoinjector or an oral version of an injectable. Companies have been known to implement hard hops (withdrawing the soon-to-be generic medicine from the market, leaving doctors with no choice but to prescribe the new version) or soft hops (keeping the old medicine on the market but not promoting it).
Mergers and Acquisitions (external innovation): When a patent cliff is too steep to fight off with legal maneuvers alone, pharmaceutical companies can use their massive cash reserves to buy out the very companies, platforms, or specific assets that can immediately replace eroding revenue. This can involve buying out smaller biotech firms with a few medicines in their pipeline (called “bolt-on acquisitions”) or a mega-merger with a large, diversified pipeline.
Altogether, the patent cliff introduces an expiration date on the comfort zone of raking in billions of sales in perpetuity. It sparks the discovery of entirely new drug classes and forces fierce innovation within existing classes. Without the patent cliff, the pharmaceutical industry would naturally stabilize into a series of permanent, highly profitable monopolies. By guaranteeing that every monopoly will expire, the system ensures that the survival of the largest healthcare companies in the world is permanently tied to the continuous, relentless pursuit of novel biology and new medicines for patients. Indeed, the patent cliff provides the strongest motivating force for new innovation in the biotech industry (aside from benefitting patients, of course). New innovation can come from within (product hopping) or from outside (M&A), which is where we come to China Biotech.
The Good: Accelerated Innovation
“My dear, here we must run as fast as we can, just to stay in place. And if you wish to go anywhere you must run twice as fast as that.” Lewis Carroll, Alice in Wonderland

Moore’s Law is an observation coined by Intel co-founder Gordon Moore in 1965 stating that the number of transistors on an integrated circuit doubles approximately every two years with a minimal increase in cost, leading to more powerful and cost-effective computing. This principle has guided semiconductor development for nearly 60 years, and characterized the exponential pace (literally) of innovation in CPU development.
The inverse law applies to biotech. In 2012 paper published in Nature Reviews Drug Discovery, the industry analyst and drug R&D consultant Jack Scannell coined Eroom’s Law (a play on “Moore’s Law” spelled backward), an observation that pharmaceutical research and development is becoming slower and more expensive over time, despite massive advances in technology. This pithy and rather unfortunate law derived emerged from Scannell’s discovery that the number of new drugs approved per billion dollars spent on R&D has halved roughly every nine years since 1950.
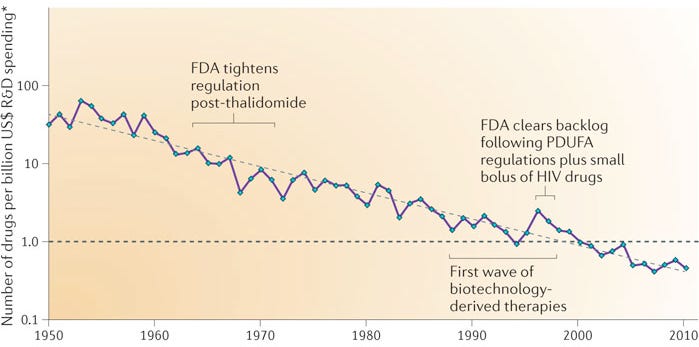
He identified four bottlenecks that may have led to this phenomenon, but the “Cautious Regulator” Problem is the one most relevant to our discussion about China Biotech (the other three are “Better-than-the-Beatles” Problem requiring new drugs to be meaningfully better than already good old drugs, “Throw Money at It” Tendency to prioritize shots on goal instead of R&D efficiency, “Basic Research Brute Force” Bias that floods the top of the funnel with false positives). The “Cautious Regulator” Problem refers to the ballooning cost and time of running clinical trials as a direct result of an increasingly risk-averse FDA. This is perhaps the single most significant structural drag on pharmaceutical R&D efficiency.
Over the decades, major public health crises (like the thalidomide tragedy in the 1960s or the Vioxx recall in 2004) have forced regulatory agencies like the FDA to systematically raise the threshold for safety and clinical data. Phase 3 clinical trials that once required a few hundred patients now routinely require thousands or tens of thousands. Every incremental increase in the regulatory safety bar adds hundreds of millions of dollars to the cost of development and drastically reduces the probability of technical success. Today, the often-cited “gold standard” figure for the average cost of developing a single new drug is roughly $2.6 billion.
However, over the last decade, China’s regulatory body for pharmaceuticals, the National Medical Products Administration (NMPA), has implemented a series of radical, top-down structural reforms that effectively solved this bottleneck, transforming China from a slow, regulatory backwater into the fastest clinical trial engine in the world.
To appreciate China’s solution, it helps to look at the three core friction points that make Western regulation so slow and expensive:
The Accumulation of Regulatory Moats: Every major pharmaceutical safety crisis in Western history has resulted in a permanent layer of new bureaucracy. The thalidomide tragedy of the 1960s mandated strict efficacy proof, while the high-profile regulatory withdrawal of Vioxx in 2004 fundamentally altered how the FDA views long-term cardiovascular risk. These rules are rarely rolled back, meaning the regulatory framework only becomes heavier, never lighter.
Exploding Trial Sizes and Timelines: To satisfy cautious regulators looking for ultra-rare safety signals, Phase 3 trial protocols have ballooned. Indication cohorts that once required 300 patients now routinely require 3,000 to 5,000 patients, drastically extending recruitment timelines.
The IND (Investigational New Drug) Administrative Backlog: In conservative systems, simply getting permission to start a Phase 1 trial involves months of paperwork, administrative stagnation, and iterative chemistry, manufacturing, and controls (CMC) audits before a single human is dosed.
Beginning in 2015, under the leadership of reformists like Bi Jingquan, China initiated comprehensive updates to address administrative bottlenecks that were impacting local clinical development timelines and delaying patient access to life-saving therapies. China’s regulatory bodies implemented a series of systematic structural updates designed to optimize clinical development timelines across four pillars:
The 60-Day Implied IND Approval (Eliminating Administrative Waiting): Historically, it took Chinese biotechs up to a year just to get approval to start a Phase 1 clinical trial. To fix this, the NMPA harmonized with Western standards but added a strict timeline constraint: if the regulator does not raise an objection or an inquiry within 60 business days of an IND submission, the trial is automatically approved to proceed. This instantly matched or exceeded the speed of the U.S. FDA’s 30-day window, cutting months of dead time out of early-stage R&D.
Achieving Harmonization via ICH Guidelines: In 2017, the region officially joined the International Council for Harmonisation (ICH). This structural alignment enabled local regulatory bodies to instantly import decades of global regulatory best practices without having to slowly iterate them internally. By aligning its clinical data requirements with global standards, China made it possible for international data to be accepted locally, and more importantly, ensured that the massive volume of clinical data generated in China’s hyper-speed ecosystem would be structured correctly for future out-licensing to Western markets.
Leveraging Massive, Centralized Patient Pools: The NMPA streamlined the institutional review board (IRB) and clinical site approval processes across China’s massive, state-run hospital networks. Since China has highly centralized medical centers that see tens of thousands of patients daily for specific oncology and immunology indications, a Chinese biotech can complete patient enrollment for a trial in a fraction of the time it takes a Western counterpart. By removing the administrative friction to activate these mega-sites, the NMPA turned China’s population scale into a structural R&D velocity advantage.
Hyper-Fast “Breakthrough” and Conditional Approvals for Cutting-Edge Modalities: The NMPA recognized that it could not beat the West in legacy small-molecule chemistry, so it created aggressive regulatory fast-tracks specifically tailored for next-generation modalities like cell therapies (CAR-T), bispecific antibodies, and antibody-drug conjugates (ADCs). For drugs targeting severe, unmet medical needs, the NMPA aggressively utilizes conditional approvals based on early Phase 2 surrogate endpoints (like overall response rate or progression-free survival in cancer), allowing drugs to reach the market and generate commercial revenue years before their final Phase 3 confirmatory data is fully baked.
By solving the “Cautious Regulator” Problem locally, China created a massive structural advantage for global biotech. Today, a drug candidate can be designed, cleared via an IND, and run through human proof-of-concept trials in China at a velocity and cost structure that is nearly impossible in the U.S.
The OK: Capturing the “Me-Better” Arbitrage
“May it be a light to you in dark places, when all other lights go out.” J.R.R. Tolkien, The Fellowship of the Ring

In the pharmaceutical industry, “me-too” and “me-better” are terms used to describe drugs that are structurally similar to an already approved, pioneering “first-in-class” drug. They target the exact same biological mechanism, but their development strategies and clinical impacts differ significantly. A “me-too” drug is a follow-on therapeutic that uses the same mechanism of action as the innovator drug and offers comparable clinical efficacy and safety profiles, with no major advantages. A “me-better” drug targets the same established mechanism but features intentional modifications to secure a clear, quantifiable clinical advantage over the pioneer in terms of safety, efficacy, or convenience. Separately, arbitrage is the practice of leveraging a price difference for the exact same asset, commodity, or service across two or more markets.
Put them together and you get a shiny new business model: the “Me-Better” Arbitrage model, where Western VCs let the streamlined NMPA framework de-risk the molecule in human patients quickly and then package that clean data into an asset ready for a potential Big Pharma acquisition. China’s hyper-efficient regulatory engine is the exact mechanism that feeds the “Me-Better” Arbitrage model. The model has evolved from an occasional creative structuring trick into a highly institutionalized, rapid-exit blueprint for cross-border drug development.
To fully grasp this phenomenon, it helps to look at the exact anatomy of an arbitrage and how it acts as a geopolitical firewall for international innovation. Unlike traditional biotech company creation, which focuses on building long-term, multi-decade platform technologies with massive brick-and-mortar footprints, a NewCo is structurally designed to be a temporary vehicle to house a specific drug. The model follows four general phases:
In-Licensing a “Me-Better”: A Western venture capital (VC) firm identifies a highly promising asset developed by a Chinese biotech. Usually, this asset has already generated early, encouraging data in local Chinese clinical trials. The Western VC licenses the “Ex-China” rights (typically encompassing the U.S., Europe, and Japan), while the Chinese firm retains domestic commercial rights.
Establishing a NewCo Shell: The VC firm spins up a lean, clean startup (the “NewCo”) often domiciled in Delaware or Europe. The company is funded with just enough seed capital to execute an immediate operational objective, such as filing a U.S. IND (Investigational New Drug) application or starting a targeted Phase 1/2 trial.
Hiring a “SWAT Team”: Instead of hiring a massive workforce, the NewCo operates with a skeleton crew of highly experienced “swat team” executives, often seasoned drug developers or entrepreneurs who have successfully sold companies before. The NewCo completely outsources its clinical trials, regulatory filings, and manufacturing scale-up to third-party Contract Research Organizations (CROs) and CDMOs. Most dollars raised go directly into driving the specific clinical asset forward.
Arbitrage Exit: Within 12 to 24 months of formation, sometimes even before the NewCo has run a single trial in the U.S., the asset’s data matures in China. If the data looks spectacular, a Big Pharma company can step in and buy the entire NewCo shell outright. If an acquisition occurs, the VCs secures a rapid and capital-efficient liquidity event, and Big Pharma takes ownership of the drug to plug into their global Phase 3 machine.
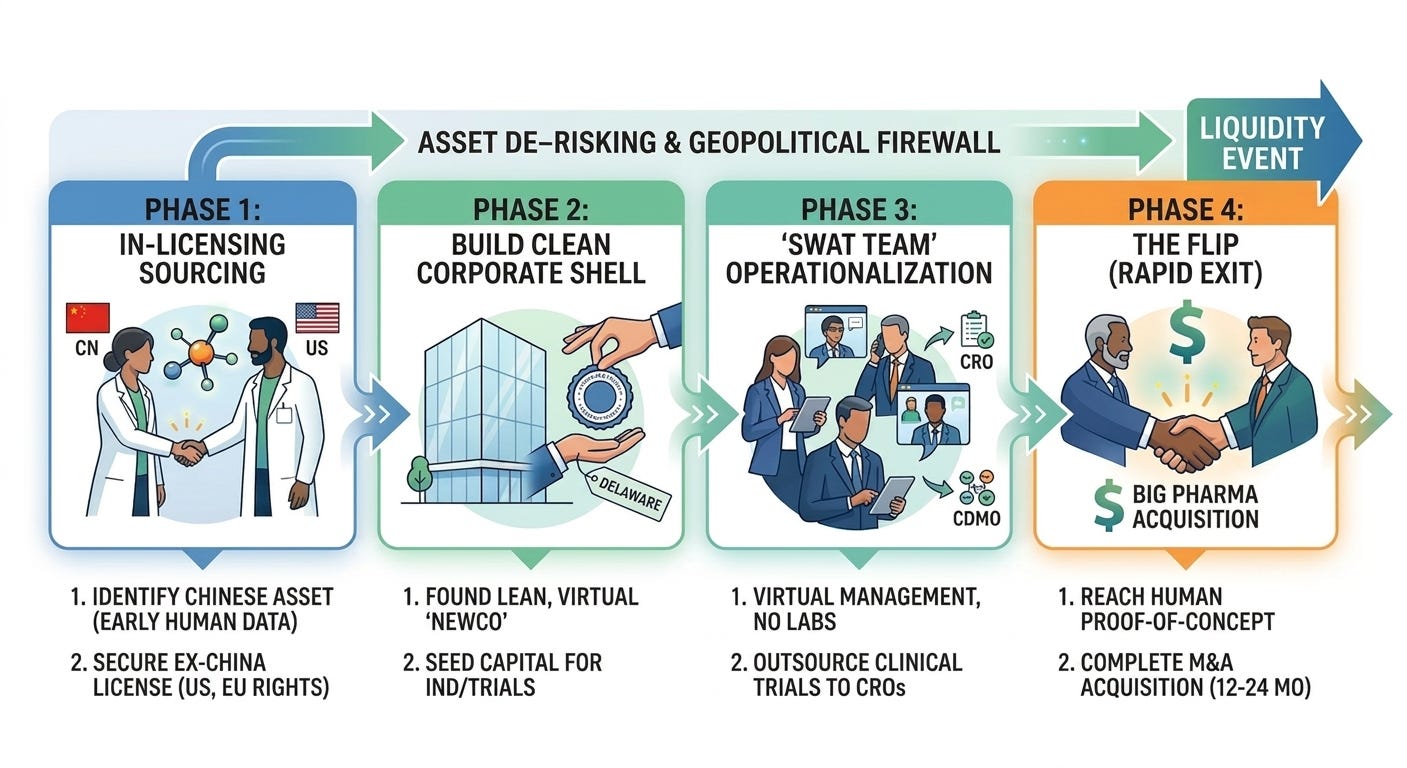
There are several reasons why this model works exceptionally well, when executed with the right expertise:
Speed to Data: Since Chinese biotechs operate at an incredibly high velocity and have access to massive patient pools, they can advance an asset from discovery to proof-of-concept in humans far faster and cheaper than a U.S. counterpart. The Western NewCo essentially buys time, skipping years of early-stage discovery risk.
Extreme Capital Efficiency: Traditional platform biotechs burn hundreds of millions of dollars maintaining wet labs, internal manufacturing, and massive corporate overhead. A NewCo typically has no brick-and-mortar labs. R&D is outsourced entirely to contract research organizations (CROs). Most dollars raised are deployed directly into advancing one or two clinical assets, maximizing the return on data generated.
Geopolitical Firewall: As U.S.-China geopolitical tensions escalated, culminating in legislative pushes like the BIOSECURE Act, Big Pharma became incredibly wary of buying Chinese companies outright or entering messy, direct cross-border partnerships that could draw scrutiny from Washington. By transferring the Intellectual Property (IP) to a clean Delaware or European corporate entity, staffed by Western executives and backed by Western VCs, the asset is effectively Westernized. This makes it politically palatable for an American or European multinational to acquire.
“Me-Better” Arbitrage model is best exemplified in these three case studies:
Candid / UCB (2026, $2 billion upfront): I covered the Candid / UCB acquisition in an Acquisitions piece, linked here.
Aiolos Bio / GSK (2024, $1 billion upfront): Aiolos Bio was founded in 2023 by industry veterans Khurem Farooq (previously CEO of Gyroscope Therapeutics acquired by Novartis), Dr. Anthony (Tony) Adamis (co-discoverer of Macugen, the very first anti-VEGF drug approved for wet AMD), and Elizabeth Holt (experienced business development and strategy leader). Aiolos Bio was launched specifically to in-license AIO-001, a long-acting anti-TSLP monoclonal antibody for asthma. The molecule was originally developed by Jiangsu Hengrui Pharmaceuticals in China, where it had already demonstrated promising early-stage clinical data. By taking a molecule that had already been clinically validated under China’s rapid NMPA framework, Aiolos bypassed the high-risk preclinical and early Phase 1 hurdles in the West. In January 2024, just months after Aiolos was publicly unveiled, GSK acquired the company for $1 billion upfront and up to $400 million in regulatory milestones.
ProfoundBio / Genmab (2024, $1.8 billion): ProfoundBio was established by ex-Seagen scientists to develop next-generation Antibody-Drug Conjugates (ADCs) targeting solid tumors. While headquartered in Seattle to interface with Western capital, its core discovery and development labs were strategically positioned in Suzhou, China, to leverage local engineering velocity and cost efficiencies. The flagship asset, rinatabart mafodotin (Rina-S), targeted folate receptor alpha (FRα) for ovarian cancer. ProfoundBio used its dual-geography footprint to rapidly generate Phase 1/2 clinical data in record time, out-pacing purely Western-siloed startups. In April 2024, antibody specialist Genmab acquired ProfoundBio for $1.8 billion in cash.
Ultimately, the “Me-Better” Arbitrage model represents a masterclass in modern risk management and geopolitical positioning. Yet, as with any highly lucrative arbitrage, its very success has signaled to the largest players in the industry that the game is changing. As multi-billion-dollar syndicates prove just how viable this direct pipeline can be, a more disruptive question inevitably emerges: if the path from local discovery to global development is now this well-mapped, how long will Big Pharma continue to pay a premium to middlemen?
The Bad: Pharma Cuts Out the Middleman
“Even as mithril was the foundation of their wealth, so also it was their destruction: they delved too greedily and too deep. You know what they awoke in the darkness of Khazad-dum...shadow and flame.” J.R.R. Tolkien, The Fellowship of the Ring

The rise of “Me-Better” Arbitrage highlights a structural shift in the biopharma industry: the decoupling of drug discovery from R&D. Historically, the companies that discovered the biology were expected to develop the drug. In the “Me-Better” Arbitrage era, China Biotech has increasingly become the discovery engine of the world, while Western venture capital and Big Pharma act as the development and commercialization engine. While this model has delivered staggering, rapid returns for venture capitalists and provided vital lifelines of non-dilutive upfront cash to Chinese innovators, it poses an existential question for the biotech ecosystem: why can’t pharmaceutical companies cut out the middleman and directly license medicines from China Biotech?
Well, they can and they will. Rather than routing single assets through NewCo shells, Big Pharma is deploying direct licensing to construct massive, multi-program portfolios with the stroke of a pen, as illustrated by a spate of recent deals:
GSK / Hengrui (July 2025): GSK bypassed external incubation to sign a massive 12-asset, up to $12 billion biobucks agreement directly with Jiangsu Hengrui Pharmaceuticals, anchored by a $500 upfront fee. The centerpiece was HRS-9821, a clinical-stage PDE3/4 inhibitor for COPD that strategically slotted into GSK’s core respiratory franchise. However, this scaled collaboration framework inherently bypassed the traditional NewCo model. GSK secured options to take over 11 additional early-stage programs in immunology and oncology once Hengrui finishes Phase 1 testing. GSK essentially secured a proprietary portfolio framework, bypassing the traditional asset aggregation timelines associated with VC syndications.
Bristol Myers Squibb / Hengrui (May 2026): In one of the broadest direct licensing plays to date, BMS committed to an astonishing $15.2 billion mega-deal covering 13 early-stage oncology and hematology programs originated by Hengrui, backed by $950 million in near-term structured payments. Rather than buying a series of individual NewCos at a high premium per molecule, BMS went straight to the source to buy an entire oncology sub-portfolio, securing an immediate, diversified influx of next-generation clinical candidates to feed its global trial infrastructure.
Pfizer / Innovent (May 2026): Pfizer entered a sprawling global strategic collaboration with Innovent Biologics to co-develop and license 12 breakthrough early-stage cancer therapies, driven by a $650 million upfront payment and up to $9.85 billion in backend milestones. The portfolio consists of eight Innovent-originated assets (including novel ADCs and multi-specific antibodies) and four Pfizer-proposed discovery assets. Instead of a VC firm flipping a shell company to Pfizer, Pfizer and Innovent cut a direct co-development deal, sharing clinical costs globally, dividing geographic commercial rights, and splitting profits in the U.S. and Europe.
AstraZeneca / CSPC Pharmaceutical Group (January/May 2026): A major limitation of the asset-centric NewCo model is that it typically isolates a single drug. Big Pharma, however, often wants access to the underlying enabling technology or unique biological modalities that Chinese biotechs have spent years optimizing. Direct licensing enables this technology transfer. AstraZeneca signed a $4.7 billion total value agreement ($1.2 billion paid upfront in May 2026) with CSPC for an 8-program weight management and metabolic pipeline. While the deal included a clinical-ready dual GLP-1/GIP agonist (SYH2082), AstraZeneca’s true prize was CSPC’s proprietary LiquidGel sustained-release delivery platform (designed to achieve once-monthly peptide dosing) and their vertically integrated, AI-driven peptide discovery engine.
These partnerships suggest that the Chinese Biotech ecosystem has evolved beyond a loose collection of early-stage startups requiring the nurturing dollars of Western venture capital. Instead, they are mature, well-capitalized innovative powerhouses capable of structuring institutional, double-digit-billion-dollar alliances directly with global multinational peers.
The Ugly: Obsolescence of U.S. Biotech
“Oh, I’m real. Real enough to defeat you! And I did it without your precious gifts, your oh-so-special powers. I’ll give them heroics. I’ll give them the most spectacular heroics anyone’s ever seen! And when I’m old and I’ve had my fun, I’ll sell my inventions so that everyone can be superheroes. Everyone can be super! And when everyone’s super...no one will be.” Syndrome, The Incredibles

Commoditization is the process by which products or services lose their distinct uniqueness and become entirely interchangeable, shifting the primary basis of competition strictly to price. Naturally, it is one of the most studied and feared forces in macroeconomics and corporate strategy.
Richard D’Aveni, a professor of strategic management at Dartmouth College’s Tuck School of Business, is recognized for introducing the concept of hypercompetition. In his 2010 book, Beating the Commodity Trap: How to Maximize Your Competitive Position and Increase Your Pricing Power, D’Aveni explains that commoditization can function as a structural shift that is actively leveraged by specific competitors. He outlines three specific commodity traps, but deterioration (the low-cost squeeze) is the one most relevant to this discussion.
Deterioration is the most common and visible form of commoditization. It starts when a low-cost entrant captures the bottom of the market. Initially, incumbent companies ignore them, assuming customers will always pay a premium for brand equity and superior features. However, as the low-cost entrant’s quality crosses the threshold of “good enough,” they being to capture market share higher up in the value chain. In this trap, market disruption doesn’t come from a technological breakthrough, but from an operational, geographical, or regulatory cost advantage. It is the exact macroeconomic engine that drove the offshoring of industries like steel, random access memory (RAM) chips, smartphones, laptops, solar cells, lithium-ion batteries, rare earths, as well as Active Pharmaceutical Ingredients (APIs) and generic drugs.
But, aren’t branded medications safeguarded by Intellectual Property (IP)? Doesn’t that prevent commoditization? Not exactly. A low-cost entrant can sometimes offer a distinct alternative to erode an incumbent’s margins without replicating the IP holder’s exact technology. They just need to create an alternative that bypasses the IP and crosses the customer’s threshold of “good enough.” A notable example of IP failing to defend against deterioration is the commoditization of personal computers (PCs).
In the early 1980s, IBM dominated the personal computer market. To block competitors, IBM heavily patented the BIOS (Basic Input/Output System), the essential chip that allowed the computer’s software to talk to its hardware. They believed this proprietary IP would permanently secure their premium market position. A competitor called Compaq hired a team of engineers who had never seen IBM’s code and used “clean-room reverse engineering” to mimic the exact functionality of the BIOS chip without copying the design.
The landmark case that legally cleared Compaq and solidified the legality of “clean-room reverse engineering” was IBM Corp. v. Compaq Computer Corp., which ultimately culminated in a private, out-of-court settlement in 1986 without an explicit judicial determination of liability. Since the IBM-Compaq dispute ended in a private settlement, the definitive legal stamp of approval for this exact methodology came a few years later in the landmark case Sega Enterprises Ltd. v. Accolade, Inc. (977 F.2d 1510). In that case, Accolade reverse-engineered Sega’s video game console BIOS to make their third-party games compatible with Sega hardware. The Ninth Circuit Court of Appeals held that disassembly and reverse engineering of copyrighted software for the sole purpose of achieving interoperability can constitute ‘Fair Use’ under the law under specific statutory conditions, provided it is the only way to gain access to those functional elements. Legally cleared of IP infringement, Compaq, Dell, and Gateway flooded the market with IBM-compatible clones. Since these clones performed nearly identically but cost a fraction of the price, IBM’s hardware pricing power evaporated, eventually forcing it to exit the PC manufacturing business entirely.
“Me-better” drugs could achieve a remarkably similar end when it comes to driving market commoditization and triggering a low-cost squeeze, though they might arrive there through a slightly different competitive mechanism than true generics. While a generic drug is an identical copy of an off-patent molecule, a “me-better” (or “me-too”) drug is a chemically distinct but functionally similar molecule launched by a competitor while the original pioneer drug is still heavily protected by patents. Even though a “me-better” drug has its own separate patent and a distinct chemical structure, insurance companies, pharmacy benefit managers (PBMs), and hospital formulary committees frequently categorize them under similar tier structures as therapeutic substitutes for reimbursement purposes.
The history of blockbusters is defined by this exact commercial dynamic. The Statin Wars in the 1970s and 1980s provide a classic case study. Merck launched Mevacor (lovastatin) as the first-in-class cholesterol-lowering statin. Pfizer fast-followed years later with Lipitor (atorvastatin). Lipitor was chemically distinct and was perceived by cardiologists as pharmacologically “better” because it was numerically more potent at lowering LDL cholesterol at lower doses. Pfizer used this slight clinical edge to gain market share, turning Lipitor into the best-selling drug in pharmaceutical history.
There are certainly barriers that prevent China Biotech companies from directly launching their “me-betters” into the United States (soft FDA mandate of domestic trials, high cost of commercialization, navigating the PBM/payor maze). However, after a few +$10 billion milestone payments, what concrete barriers truly prevent China Biotech from directly competing in the U.S. market?
Even if China Biotech doesn’t directly compete in the U.S. market, and continues to license post-proof-of-concept drugs into pharma pipelines, what is the residual value of U.S. biotech companies? How can U.S. biotech compete and afford to hire a workforce when their niche (the legacy innovation engine for pharma pipelines) is undercut by a highly skilled, highly motivated, lightning fast competitor?
These are questions that the biotech industry will have to answer in the coming years (or earlier).
How Do We Protect Our Biotech Crown Jewel?
The COINS Act, officially known as the Comprehensive Outbound Investment National Security Act, is a landmark piece of federal legislation that codifies, expands, and hardens the United States’ framework for regulating outbound investment (the flow of American capital, joint ventures, and technology transfers into foreign countries). The bill was signed into law on December 18, 2025, as a central component of the Fiscal Year 2026 National Defense Authorization Act (NDAA).
The primary objective of the COINS Act is to regulate outbound investment flows involving sensitive technologies to designated countries of concern, as defined by the U.S. Federal Government. The law directs the U.S. Department of the Treasury to implement regulations that divide outbound transactions into two clear buckets:
Prohibited Transactions: Absolute bans on U.S. persons or their controlled foreign subsidiaries investing in, joint-venturing with, or directing capital toward entities in a designated country of concern that develop specific high-level, dual-use technologies.
Notifiable Transactions: Mandatory reporting requirements for investments in lower-threshold or adjacent technology tiers. These require firms to submit detailed disclosures to the Treasury Department, allowing the government to track technology diffusion and build enforcement data.
The COINS Act originally preserved restrictions on the three core technology sectors, but was expanded to add new sectors (see table below).
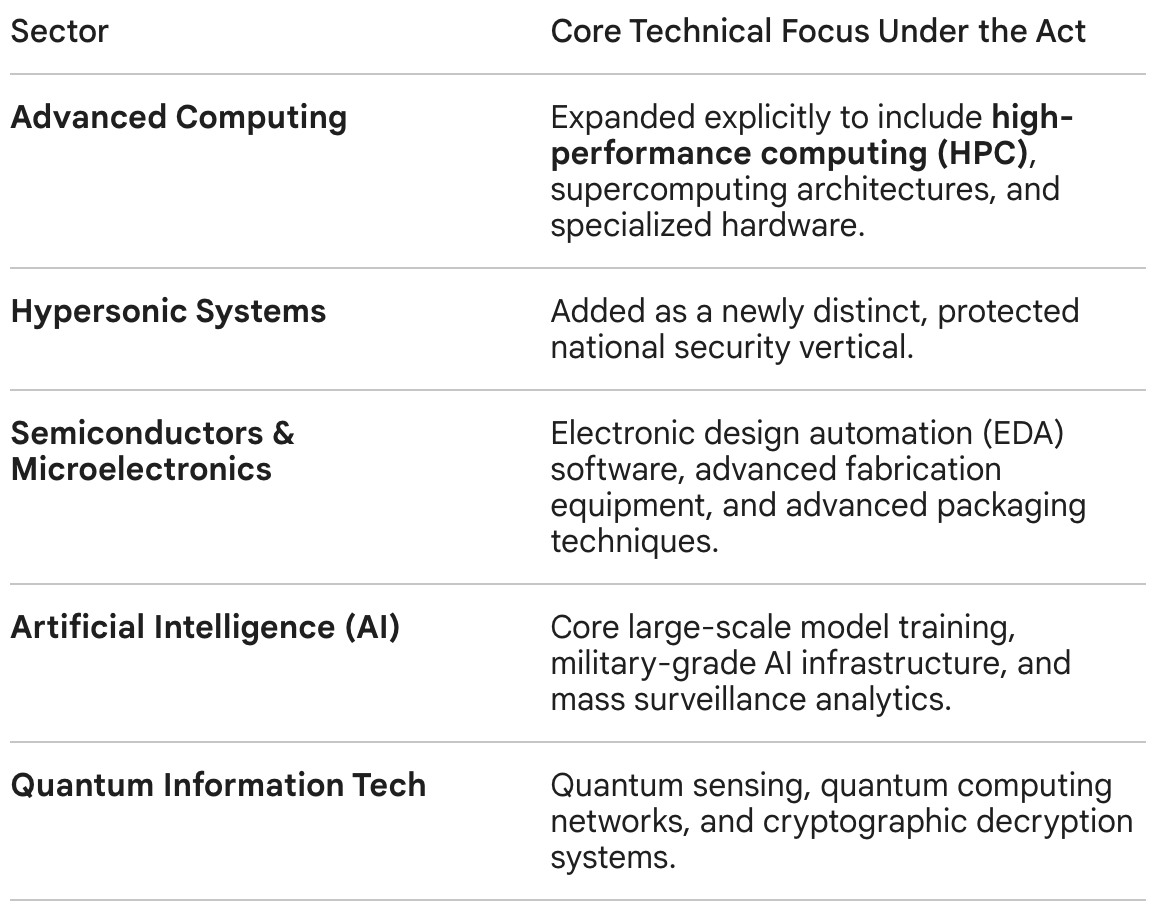
Reflecting a sharp bipartisan shift in early 2026 regarding bio-industrial independence, lawmakers have proposed the integration of advanced biotechnology, genomics infrastructure, and proprietary drug licensing platforms into the COINS Act framework. This could block U.S. firms from executing cross-border licensing or early-stage co-development deals with advanced therapeutic developers in designated countries of concern. One of the most contentious points of discussion in the debate on China Biotech is whether or not biotechnology should be included in the COINS Act.
The “hawks” see biotechnology as a critical geopolitical technology that should be included in the Act. Led heavily by figures like Chairman John Moolenaar of the House Select Committee on the Chinese Communist Party (CCP), the argument for adding biotech to the COINS Act framework treats advanced medicine and genomics not just as healthcare, but as dual-use national security assets. Proponents of the expansion point to a staggering trend: in 2025, cross-border out-licensing deals between Western pharma giants and Chinese biotech firms reached $136 billion, making up nearly half of all major global pharmaceutical licensing transactions. Massive co-development deals are viewed by hawks as a potential mechanism for the transfer of Western intellectual property know-how, which they argue could accelerate regional advancements up the pharmaceutical value chain.
On the other side of the aisle, life science venture capitalists and legal trade groups are warning that over-regulating outbound capital flows in life sciences could paralyze drug development. Unlike semiconductors, which are highly localized, modern biotechnology relies on a deeply interconnected, cross-border funding ecosystem. Many early-stage U.S. biotechs rely on international joint ventures and global co-development partnerships to finance multi-million-dollar clinical trials. Industry groups argue that broadly categorizing biotechnology as ‘prohibited’ under the COINS Act could increase transaction friction, asserting that if routine out-licensing structures require Treasury notification or face restrictions, deal teams may find it increasingly difficult to underwrite the associated regulatory risk. This regulatory burden could stall or completely kill cross-border clinical trials, ultimately delaying the commercialization of novel, life-saving oncology or metabolic therapies in the West. Industry advocates are pushing back against a blanket ban, arguing that if Treasury must include biotech, the rules should be narrowly tailored. They argue that restrictions should apply strictly to highly sensitive infrastructure (like advanced synthetic biology platforms, gene-editing tools (CRISPR), or mass genomic data harvesting) while exempting conventional small-molecule or monoclonal antibody drug development.
The debate is further complicated by the fact that Washington has already targeted Chinese biotech from the inbound/procurement side. The FY 2026 NDAA successfully codified the BIOSECURE Act, which restricts U.S. federal agencies from contracting with or funding entities designated as ‘Biotechnology Companies of Concern’ (such as WuXi AppTec or BGI). The push to add biotech to the COINS Act is viewed by some as the necessary “second half” of this strategy.
As the Treasury Department moves through its formal notice-and-comment rulemaking period, with final implementations statutorily mandated to go into effect by March 2027, the life sciences sector is facing unprecedented scrutiny. While the possibility of a blanket ban on all pharmaceutical collaboration with China is strained due to the intense blowback from patients and industry groups, industry analysts expect the Treasury to institute mandatory notification requirements for high-value intellectual property transfers, co-development frameworks, and joint ventures in designated countries of concern. For biopharma dealmakers, the era of evaluating transactions purely through a clinical and commercial lens could be over. Every major deal might have to navigate a stringent national security framework.
Or perhaps there is a more nuanced frame, like the one that Daphne Zohar (CEO of Seaport) proposes; one in which the biotech industry collaborates with Congress to address their valid concerns, and propose constructive upgrades to U.S. biotech infrastructure (instead of impulsively attacking it like the biotech industry did with the Inflation Reduction Act, which backfired for the industry and resulted in what amounts to an accelerated generalization of top selling Medicare drugs every year).


Conclusion
“It’s a dangerous business, Frodo, going out your door. You step onto the road, and if you don’t keep your feet, there’s no knowing where you might be swept off to.” J.R.R. Tolkien, The Fellowship of the Ring

The evolution of China Biotech from a regional regulatory backwater into a high-velocity global innovation engine has disrupted the traditional pharmaceutical playbook. As we have explored through these four lenses, the phenomenon is neither purely a triumph nor a crisis.
The Good & The OK demonstrate how structural NMPA reforms and the “Me-Better” Arbitrage model have successfully de-risked early-stage molecules, offering Western capital and Big Pharma an unprecedented, highly efficient pipeline to outrun the crushing weight of impending patent cliffs. The Bad & The Ugly reveal the friction points of this transition, exposing how direct licensing deals threaten to bypass Western biotech middlemen entirely, triggering a low-cost squeeze that could ultimately commoditize legacy drug discovery infrastructure.
Today, we find ourselves at a critical crossroads. The signing of the COINS Act and the ongoing regulatory notice-and-comment period through March 2027 signal that Washington D.C. no longer views biotechnology through a purely clinical lens, but as a critical battleground for national security and bio-industrial independence.
Ultimately, trying to completely untangle a deeply interconnected, cross-border life sciences ecosystem risks paralyzing the very innovation required to cure complex human diseases. The challenge for biopharma dealmakers and policymakers alike will not be deciding whether to engage with China Biotech, but learning how to safely navigate this new geopolitical firewall. In the relentless race against biology and patent expirations, the winners may not be those who build the highest walls, but those who learn to run the fastest in a hypercompetitive world.
Love biotech? Check out Biotech Readout’s full content library, or navigate directly to a segment that interests you:
Frontiers in Medicine: Exploring the frontiers of our understanding and treatments for disease.
Medical History: Recovering forgotten relics in the history of medicine.
Acquisitions: Exploring the innovation behind acquired companies.
Weekly Readout: A digest of new clinical data from the past week.

Epilogue
In this piece, I did not aim to push either “for or against” a deeper engagement with China Biotech but, rather, attempted to lay out diverse viewpoints and their consequences. To further improve balance, I am listing a few outside perspectives that I found particularly informative and are either more enthusiastic or more cautious about deeper U.S.-China engagement:
More enthusiastic about deeper U.S.-China engagement:
John Crowley (CEO of BIO) says, “Capital is stateless, rational, and fluid […] It would be a fool’s errand to try to stop what’s happening in China.”
Robert Zirkelbach (PhRMA’s chief public affairs officer) is against adding biotech to the COINS Act and says", “We’re not going to regulate ourselves to success here”
John Maraganore (founder and former CEO of Alnylam) says, “We are the greatest country for DISRUPTIVE innovation: […] China is emerging as a source of INCREMENTAL innovation. It’s great for patients to have BOTH, and we should put patients FIRST as our Northstar.
Bruce Booth (Parter at Atlas Venture) says, “I’m firmly in the camp that their engagement in the global ecosystem is good for the sector”
Roderick Wong (CIO & Managing Partner at RTW) says, “By company creation, slightly more than the majority [of pipeline programs in 2026] would come from China […] By dollar value, it’ll end up being more to China.”
Roger Perlmutter (CEO of Eikon Therapeutics, former EVP of Merck & Co. and former president of MRL) says China is investing in science while U.S. reducing investment and, in that environment, we shouldn’t be surprised if China Biotech starts to outperform U.S. Biotech
Anonymous investor who has built startups around Chinese-invented drugs interviewed by STAT News says, “The uncomfortable reality is when these things happen, there are winners and losers. […] That’s table stakes for full-contact American capitalism.”
Anonymous biotech investor interviewed by STAT News says, “Until I’m not allowed to do it, of course I’m going to do it. [...] There are too many for-profit actors in this setting who are willing to launder their ethics to make $100 million on each deal.”
C. Simone Fishburn (Editor in Chief at Biocentury) and Joshua Berlin (Head of BD at Biocentury) say, “The ability of U.S. biotech executives, policymakers and some investors to deny the rising innovation and inventiveness from China is astonishing. But no degree of protectionism, and no willing it away, is going to change the reality that the U.S. is facing ever-increasing competition from a new geographic center of excellence”.
More cautious about deeper U.S.-China engagement:
John Moolenaar (Congressman and chair of the House of Representatives’ China committee) says, “I write to draw your attention to a dangerous surge of American capital and know-how into [China’s] biotechnology sector […] I urge [the] Treasury to give particular consideration to transactions involving the licensing of pharmaceutical intellectual property.”
Jason Kelly (Co-founder and CEO of Ginkgo Bioworks, Former Chair of US National Security Commission on Emerging Biotechnology) says “Put biotech on COINS Act list of strategic technologies and can slow this down easily. Common sense and good for US science and patients.” Thank you for the shoutout Jason!


Scott Gottlieb (Senior Fellow at AEI, Partner at NEA, and former Commissioner of the FDA) says “China’s new rules require approval before Chinese tech is transferred overseas. But U.S. drug tech is open for Chinese firms to parse for molecular leads that are quickly turned into competing medicine. Knowledge flows in one direction: into China not out.”
Dr. Robert Califf (former Commissioner of the FDA) says, “I think the concerns are valid and very real. […] The U.S. is being seriously threatened.”
Curie Bio (biotech investment fund) has been circulating a document among lawmakers about Chinese biotechs titled “China is running its rare earth playbook in the new medicine supply chain”.
Olivia Kosloff (senior fellow at American Economic Liberties Project) says, “Pharma and biotech leaders are destroying their own industry in pursuit of short-term profits. Companies are growing too dependent on China. […] Instead of throwing over American companies in pursuit of short-term financial gains, American pharmaceutical leaders should be advocating for policies that will protect American biotech: reform of the clinical trial ecosystem to make it less expensive to bring a drug to market, preferential investing in American assets, and even the addition of Chinese assets to the COINS Act, which prevents American investments in sensitive industries in countries of concern.”
Brad Loncar (founder of Biotech TV) says, “When I launched my China biotech fund on Nasdaq eight years ago, literally nobody wanted to hear about it. Now China is all anybody talks about here. […] I do believe the U.S. needs to act in some way. It’s fine and good to say patients first, but the bigger picture gets dangerous if our country outsources too much science to a country that is, if we are being honest, an adversary.”
Anonymous CEO of U.S. biotech company interviewed by STAT News says, “They get you dependent on something and then they use it as leverage. […] They’re the coke dealers of globalism.”
Anonymous investor interviewed by STAT News says, “My belief is our entire [biotech] industry is dead in 10 years if things don’t change, so why not [block U.S. investments in Chinese biotech]?”
Joe Lonsdale (Managing Partner at 8VC and co-founder of Palantir Technologies, Addepar, and OpenGov) says, “China is not “competing” with American biotech, this isn’t a level playing field. The Chinese Communist Party (CCP) is flooding its companies with capital, tilting rules for domestic champions, and leaning on IP theft and coercion. To fix this, the next leader of the FDA must be a fighter. This includes not just embracing innovation but also confronting and at times overruling the internal bureaucracy. The next commissioner can do all of this by prioritizing speed, domestic resiliency, and regulatory clarity across drugs and clinical AI.”
To contact us, please send us an email at biotechreadout@gmail.com
Disclaimers
Investigational Status Disclaimer
The therapeutic candidates discussed in this newsletter are currently in clinical development and have not been approved for commercial sale by the U.S. Food and Drug Administration (FDA), the European Medicines Agency (EMA), or other global regulatory authorities. Their safety and efficacy have not been established. References to pipeline products and ongoing clinical trials involve significant risks and uncertainties. Statements regarding the potential safety, potency, or efficacy of investigational drugs reflect current hypotheses and are not a guarantee of future performance or regulatory clearance. The outcome of clinical trials is inherently unpredictable, and clinical results from earlier stages may not be predictive of results in later, larger-scale trials.
No Medical Advice Disclaimer
This newsletter is for informational and educational purposes only. The content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or another qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read in this publication.
No Patient-Provider Relationship Disclaimer
The information provided in this newsletter is for educational and analytical purposes only. Receipt of this information, or any interaction with this content, does not create a physician-patient, pharmacist-patient, or any other professional-provider relationship between you and the authors or publishers. This newsletter should not be used as a substitute for a personal consultation with a qualified healthcare professional.
Forward-Looking Statements Disclaimer
This newsletter contains “forward-looking statements” regarding future events, including clinical trial timing, regulatory milestones, and projected market performance. These statements are based on current expectations and assumptions that are subject to significant risks and uncertainties. Actual results may differ materially from those expressed or implied. We undertake no obligation to update these statements as a result of new information or future developments.
Third-Party Links & Content Disclaimer
This newsletter contains links to third-party websites, including clinical trial registries and corporate presentations. Biotech Readout does not endorse, guarantee, or assume responsibility for the accuracy or reliability of any information offered by third-party providers.
Errors and Omissions Disclaimer
While we strive for technical accuracy, the information in this newsletter is provided on an “as is” basis with no guarantees of completeness, accuracy, or timeliness. Biotech Readout assumes no liability for any errors or omissions in the content of this publication.
Non-Endorsement Disclaimer
Any reference to specific commercial products, processes, or services by trade name, trademark, or manufacturer does not constitute or imply an endorsement or recommendation by the author. All trademarks are the property of their respective owners.
No Investment Advice Disclaimer
This newsletter is for informational purposes only and does not constitute financial, investment, or legal advice. The author is not a registered investment advisor. You should consult with a professional financial advisor before making any investment decisions. The biotechnology sector is highly volatile; past performance is not indicative of future results.
Conflict of Interest Disclaimer
The author of this newsletter maintains a position of independence. At the time of publication, the author holds no direct financial interest, equity, or options in any of the companies mentioned in this report. No compensation has been received from any third party to feature or analyze specific therapeutic candidates or corporate entities.