
CAR-T, Part 2
Beyond autologous CAR-T: allogeneic, in vivo, and T-cell engagers (TCE)

Disclaimer: This newsletter is for educational and informational purposes only and does not constitute medical, investment, or financial advice, nor does it establish a provider-patient relationship. Content may include forward-looking statements and discussions of investigational therapeutic candidates that are not FDA/EMA approved; their safety and efficacy remain unestablished and clinical outcomes are unpredictable. While we strive for accuracy, all information is provided as is without guarantees. This newsletter is independent, and the author holds no financial positions in the companies mentioned nor receives third-party compensation for this coverage. Revenue run rates are estimates based on the author’s synthesis of publicly available 10-K/10-Q filings and may not reflect GAAP-certified annual totals. Please find a complete version of our disclaimers at the bottom of this article and on our About page.

Introduction
In Part 1 of this series of CAR-T, we traced the roots of immunotherapy back to Coley’s Toxins, re-lived the Immunology Civil War and its détente with the 1908 Nobel Prize, unraveled the key discoveries that would enable CAR-T (T-cell and TCR), explored the progression from the proto-CAR-Ts of Dr. Yoshikazu Kurosawa and Dr. Zelig Eshhar to the long-liver costim powerhouses crafted by Dr. Michel Sadelain and Dr. Carl June, and lingered a bit on the heartwarming story of Emily Whitehead (the first pediatric CAR-T patient). Lastly and perhaps most importantly, we touched on two critical problems with CAR-T:
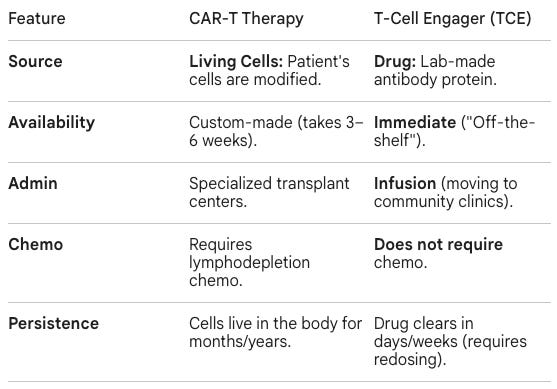
Access Gap: The month-long logistical obstacle course required to draw T-cells, re-program them, expand them, QC them, and re-infuse them back into a patient. This technically complex process largely restricted access to CAR-Ts to elite academic medical centers like UPenn.
Lymphodepleting Chemotherapy: The process of existing T-cells to make room for new CAR-T cells to multiply adds a significant layer sof treatment burden to an already frail patient.
While these barriers remain for “autologous” CAR-T therapy (when a patient’s own T-cells are re-programmed), there are a number of emerging technologies that are specifically designed to be “off-the-shelf” with the following benefits in mind:
Donor-Derived: Instead of using the patient’s own T-cells, which may be “exhausted” from prior chemotherapy, these cells are harvested from healthy, screened donors.
Ready for Use: Because the product is pre-manufactured and stored, there is no “vein-to-vein” wait time (the 2–4 week period required to engineer a patient’s own cells).
Scalability: A single donor’s leukapheresis material can potentially be used to create hundreds of doses, moving the process toward a more traditional pharmaceutical manufacturing model.
In Part 2, we will explore those therapies that push the frontier beyond autologous CAR-T:
Allogeneic CAR-T / scroll to section ⬇️
In vivo CAR-T / scroll to section ⬇️
T-cell engagers (TCE) / scroll to section ⬇️
Stay tuned for Part 3 of this three-part Frontiers in Medicine series on CAR-T, where we discuss the re-tooling of CAR-T against autoimmune diseases.
Allogeneic CAR-T, an “Off-the-shelf” Approach
Before we dive into the history here, it’s important to demystify a bit of industry jargon define the difference between “autologous” and “allogeneic” CAR-T. The fundamental difference between autologous and allogeneic therapy lies in the source of the cells:
Autologous, Self to Self: In autologous therapy, the patient is their own donor. Cells are collected from the patient via apheresis, sent to a manufacturing facility to be genetically engineered (in the case of CAR-T), expanded in number, and then infused back into the same patient. Since the cells are self, they will not attack the patient’s healthy tissues. These cells can survive for years in the body as living drugs.
Allogeneic, Donor to Patient: In allogeneic therapy, cells are harvested from a healthy, third-party donor. Cells are collected from a healthy volunteer, engineered to be universal (often by removing the T-cell receptor to prevent graft-versus-host disease), and are frozen. Healthy donors typically have more robust, fitter immune cells than heavily pre-treated cancer patients. This process is, theoretically, more scalable, allowing one donor collection to potentially provide hundreds of doses, significantly lowering costs. These doses are kept in cell banks at hospitals. Patients can be treated within days of diagnosis, as opposed to the months-long vein-to-vein time of autologous CAR-T.
The story of allogeneic CAR-T begins with André Choulika, a researcher in the laboratory of Bernard Dujon at the Pasteur Institute. His work focused on meganucleases, naturally occurring enzymes that can recognize and cut specific DNA sequences. Choulika was among the first to prove that these nucleases could be used to induce homologous recombination (HR) in mammalian cells. In 1995, his paper on I-SceI meganucleases demonstrated that one could create targeted double-strand breaks (DSBs) at a specific coordinate to trigger the cell’s natural repair machinery, potentially enabling the precise deletion or insertion of DNA. Choulika’s predated the more modern CRISPR technology by 17 years (when Jennifer Doudna’s landmark paper was published in 2012). Before Choulika’s discovery, genome modification was largely done by random integration (sticking DNA into the genome and hoping it landed somewhere useful). His paper showed that induced homologous recombination was approximately two orders of magnitude (100x) more frequent than spontaneous recombination, making precise gene editing a viable therapeutic tool rather than a rare laboratory fluke. This specific snip-and-insert logic is exactly what is used today to remove the T-cell Receptor (TCR) from donor cells.
While the Dujon lab was focused on basic biology, Choulika was focused on application. He recognized that if you could make these yeast enzymes work in human cells, you could rewrite the human genome. In 1999, André Choulika spun Cellectis out of the Pasteur Institute, making it the first company in the world dedicated specifically to genome engineering. Choulika’s first major move was securing the exclusive licenses for meganucleases from the Pasteur Institute. Initially, Cellectis didn’t focus on drugs. They functioned more like a tool shop, licensing their meganuclease technology to large pharmaceutical companies and agricultural firms who wanted to modify cell lines or crops.
By 2011, Cellectis faced a crossroad. Meganucleases were hard to engineer for new targets. Each meganuclease is a single protein that both “recognizes” and “cuts” a specific DNA sequence. If you wanted to cut a different part of the genome, you had to redesign the entire protein, a process that was slow, expensive, and often failed. Recognizing that meganucleases were too rigid for mass-scale drug development, Choulika made a aggressive strategic pivot:
Acquiring TALENs: He secured the rights to TALEN technology (Transcription Activator-Like Effector Nucleases) from the University of Minnesota and Iowa State University. Unlike meganucleases, TALENs were modular. Theoretically, you could snap different codes together to target almost any DNA sequence.
Betting on CAR-T: Instead of just being a tool company, Cellectis decided to become a therapeutics company. Choulika realized that the killer application for gene editing wasn’t just fixing a broken gene, but engineering donor T-cells to be universal.
Scientists at Cellectis worked tirelessly to engineer allogeneic “off-the-shelf” CAR-T, which they called UCART (Universal Chimeric Antigen Receptor T-cell). These cells are gene-edited to be compatible with any patient, and get around non-self immune rejection. The two key ingredients of their allogeneic CAR-Ts came from different biological threads:
TRAC (The Safety Switch): The discovery that the T-cell Receptor (TCR) was the culprit behind Graft-versus-Host Disease (GvHD) was well-established in bone marrow transplant science. However, the specific discovery that you could delete the TRAC gene to prevent GvHD in CAR-T was further validated by researchers like Hiroshi Torikai and his team at MD Anderson. In early 2012, they demonstrated that using Zinc Finger Nucleases (ZFNs) to disrupt TRAC did not hurt the T-cell’s ability to kill cancer but did stop it from attacking the host.
CD52 (The Survival Strategy): The use of Campath/Lemtrada (an anti-CD52 antibody) was already common in oncology and transplants to clear out a patient’s immune system. The discovery here was the clinical observation that Campath/Lemtrada would also kill any donor CAR-T cells. Cellectis researchers realized they could use their gene-editing tools to delete the CD52 target from the donor cells, effectively giving them a bulletproof vest against the patient’s chemotherapy.
By 2012, Cellectis brought these external concepts together and applied their proprietary TALEN technology to create a single, efficient manufacturing process. Cellectis scientists, led by Philippe Duchateau and Laurent Poirot, engineered TALENs to precisely target both the TRAC and CD52 loci in a single manufacturing run. A critical, often overlooked part of the discovery was the acquisition of CytoPulse in 2010. Its PulseAgile technology allowed Cellectis to shock the cells just enough to let the TALEN mRNA inside without killing the delicate donor T-cells. Between 2011 and 2012, Cellectis moved from theoretical editing to showing that these double-knockout cells could still proliferate, survive freezing/thawing, and kill tumor cells in mouse models.
In June 2014, Pfizer and Servier (a major French pharmaceutical firm) partnered with Cellectis in one of the largest early-stage biotech collaborations in history. Cellectis received an upfront payment of $80 million, but the total deal value exceeded $2.9 billion in potential milestone payments. Pfizer underscored its long-term commitment by purchasing a 10% equity stake in Cellectis. Pfizer secured the rights to develop allogeneic therapies for 15 oncology targets, while Servier and Cellectis shared rights to another 12 targets, including the flagship candidate, UCART19 (an allogeneic CAR-T that targets CD19).
With manufacturing and big pharma partnerships in place, UCART19 headed into the clinical trials becoming the first allogeneic CAR-T therapy ever used in a human. One-year-old Layla Richards became the first person in history to receive those gene-edited universal CAR-T cells. Layla had been diagnosed with an exceptionally aggressive form of B-cell Acute Lymphoblastic Leukemia (B-ALL) at just three months old. When all standard therapies (chemotherapy and a bone marrow transplant) failed, her lead clinician at Great Ormond Street Hospital (GOSH), Professor Paul Veys, and the medical team informed her parents that they had run out of conventional options and suggested palliative care. However, they mentioned that a highly experimental “off-the-shelf” treatment was being developed in the hospital’s own labs by Dr. Waseem Qasim, a Professor of Cell and Gene Therapy at University College London (UCL).
Rather than accepting palliative care, Layla’s father, Ashleigh Richards, specifically asked the doctors to pursue the experimental treatment. He is quoted as saying:
“She was sick and in lots of pain so we had to do something... We didn’t want to accept palliative care and so we asked the doctors to try anything for our daughter, even if it hadn’t been tried before.”
Ashleigh Richards, father of Layla Richards, the first person in history to receive allogeneic CAR-T therapy
Once the family gave their consent and expressed their desire to fight, Dr. Waseem Qasim took the initiative to bridge the gap between the lab and the bedside. Dr. Qasim contacted Cellectis (the biotech company that owned the technology) to request a special “compassionate use” license for the single vial of UCART19 that was sitting in the hospital’s freezer. He then led the effort to present the case to an emergency ethics committee to gain legal permission to use a drug that had never been tested in humans. The drug had never been tested in humans, but given Layla’s terminal status, the UK regulatory authorities granted a one-time exception.
Layla’s treatment was performed under a “Special Therapy” regulation at Great Ormond Street Hospital (GOSH). The procedure was technically complex and required a sequence of specific clinical steps:
Lymphodepleting Chemotherapy: Before the infusion, Layla was given a specific chemotherapy regimen including Campath/Lemtrada. This was intended to wipe out her remaining immune system to create a niche for the donor cells to grow without being immediately rejected.
The Infusion: Layla received a 1mL dose containing approximately 5 million engineered T-cells per kilogram of her body weight. The infusion itself took only 10 minutes.
The “Wait and See” Period: Because the TRAC gene was knocked out, doctors were watching specifically for any signs of Graft-versus-Host Disease (GvHD). If the engineering failed, the cells would have attacked her skin, liver, and gut.
The outcome was described by her lead clinician, Professor Paul Veys, as “almost a miracle.” Within 28 days of the infusion, tests showed that the UCART19 cells had successfully expanded in her blood and she was in molecular remission (no detectable cancer cells). About two weeks post-infusion, Layla developed a mild skin rash. While usually a scary sign of GvHD, in this context, it was a positive clinical marker that the donor cells were active and taking in her body. The most critical outcome was that the UCART19 cells cleared the cancer long enough to allow her to receive a second bone marrow transplant from a different donor. This transplant was necessary to provide her with a long-lasting, healthy immune system, as the UCART cells were designed to eventually disappear. Layla’s case study has since been documented in documented in several high-impact journals:
Qasim, W., et al. Science (2017): This is the definitive paper that details her specific dosage (4.6 x 10⁶ cells/kg), the “Double Knockout” (TRAC and CD52), and the molecular confirmation that the donor cells successfully eliminated her leukemia without causing GvHD.
Benjamin, R., et al. Lancet (2020): This publication expanded on Layla’s success, showing that the UCART19 platform worked in a larger cohort of 21 patients (7 children and 14 adults). It confirmed a 67% complete response rate in the pediatric group, directly tracing back to the protocol used for Layla.
With the clinical success of Layla Richards, Cellectis and their partners re-architected the future of UCAR19. In 2018, Cellectis’ partner Pfizer decided to shift its oncology strategy. While they remained interested in the technology, they felt the allogeneic portfolio would advance faster within a more agile, dedicated startup environment. Pfizer contributed its entire portfolio of allogeneic CAR-T assets (including the UCART19 rights and 16 preclinical targets) to a newly formed company: Allogene Therapeutics. In exchange for these assets, Pfizer took a 25% ownership stake in Allogene, allowing them to benefit from the upside of the technology without managing the day-to-day R&D. Allogene was founded by Arie Belldegrun and David Chang, the same team that built Kite Pharma and successfully developed the first autologous CAR-T (Yescarta). Their expertise in taking cell therapies through the FDA was seen as the missing piece to move the UCART platform from academic proof-of-concept into a commercial product. Allogene launched with $300 million in Series A funding, ensuring the UCART19 trials (CALM and PALL) would be fully funded without Cellectis having to bear the cost. Cellectis remained eligible for up to $2.8 billion in milestone payments plus royalties.
On April 13, 2026, Allogene announced that the pivotal Phase 2 ALPHA3 trial evaluating UCAR19 (now called cema-cel) versus standard of care as a first-line consolidation therapy for patients with high-risk Large B-cell Lymphoma (LBCL) showed a positive interim signal (press release, slide deck). Approximately 58.3% of cema-cel-treated patients achieved minimal residual disease (MRD) clearance at Day 45, compared to 16.7% MRD clearance among patients in the observation arm. Furthermore, patients receiving cema-cel saw a median 97.7% decrease in plasma ctDNA by Day 45, while the observation arm saw a median increase of 26.6%. In oncology, MRD (Minimal Residual Disease) refers to the small number of cancer cells that remain in the body after treatment. These cells are below the limit of detection (invisible) to standard imaging like PET or CT scans but can be detected through highly sensitive liquid biopsies that track circulating tumor DNA (ctDNA). Achieving MRD clearance is a critical clinical milestone because it indicates that the treatment has eliminated the disease at a molecular level, significantly reducing the risk of a future relapse.

Did cema-cel deliver the logistical gains that were the hope for allogeneic CAR-T? While this trial didn’t compare cema-cel directly against autologous CAR-T, there are some initial observations that we can make:
Outpatient Management: The primary barrier to outpatient CAR-T is the risk of Cytokine Release Syndrome (CRS) and ICANS (neurotoxicity). The FDA package insert and registration trials for the autologous CAR-T Yescarta reported a 94% incidence of CRS in patients with Large B-cell Lymphoma. Clinical guidelines from institutions like MD Anderson (the CARTOX algorithm) and systematic reviews in PMC confirm that patients are typically monitored inpatient for 7–14 days due to the median onset of CRS occurring within the first week (typically Day 2 to Day 5). On the other hand, allogeneic CAR-T cema-cel reported 0% CRS and 0% ICANS in the outpatient cohort, enabling 83% of patients (10 out of 12) to be managed entirely as outpatients, never requiring a hospital bed for treatment-related issues.
Community Access: A 2025 report from the University of Kansas Cancer Center, which notes that only 20% to 30% of eligible patients eventually receive therapy. This is primarily because they cannot travel to or stay near a specialized urban transplant center for the required month of monitoring. In contrast, approximately 33% of screening and infusions of allogeneic CAR-T cema-cel took place at community cancer centers, highlighting the potential for this “off-the-shelf” therapy to be administered outside of specialized academic hospitals. By removing the requirement for a 24/7 inpatient oncology ward, the potential addressable market for cema-cel could potentially be significantly larger than for any autologous product currently on the market.
Following the positive interim futility analysis reported in April 2026, Allogene Therapeutics’ next steps are focused on completing the clinical evidence required for a Biologics License Application (BLA), a comprehensive request submitted to the FDA for permission to market a new biological product in the United States. While the interim data focused on MRD clearance (molecular evidence of cancer detection), the FDA requires longer-term clinical outcomes for approval. Allogene expects to present this data on Event-Free Survival (EFS), the ALPHA3 trial’s primary endpoint, in min-2027. If successful, it could support a BLA filing and, if it clears the FDA’s stringent regulatory review process, cema-cel could become the first ever approved allogeneic CAR-T therapy.
In Vivo CAR-T, Programming T-cells in the Body
Allogeneic CAR-T offered a potential solution to autologous CAR-T’s logistical burden by allowing oncologists to delivering pre-engineered cells from “off-the-shelf” instead of triggering a month-long process of programming the patient’s own T-cells. However, it still requires lymphodepleting chemotherapy, a grueling process for already an already frail patient. In vivo CAR-T, where CAR-T cells are programmed within the body, is a potential solution to both problems.
While this technology is immature (most are preclinical or Phase 1 stage), there had been a spate of acquisitions in the space, totaling nearly $15 billion in deal value over the last 18 months (see table below). Biotech Readout discussed one of these acquisitions, Eli Lilly’s purchase of Kelonia for $3.25 billion and up to $7 billion including clinical, regulatory and commercial milestone payments. This signals unusually high enthusiasm for in vivo CAR-T among big pharma acquirers, who typically prefer to acquire FDA-approved or Phase 3 assets (~61% of acquisitions in our internal dataset) because they generate a better risk-adjusted return on investment, a must-have in order to compensate for impending patent cliffs.
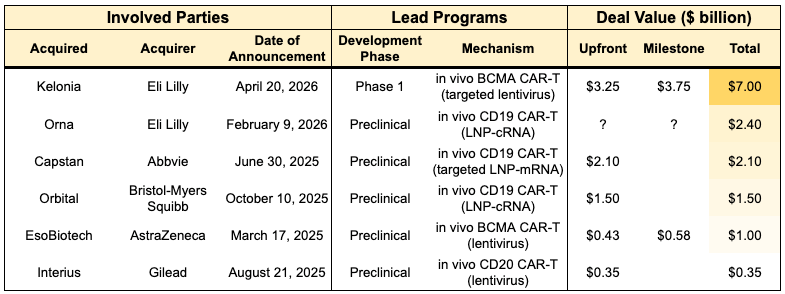
So, how do these companies aim to subvert a long and expensive manufacturing process, and instead create CAR-T within patients themselves? They are undertaking two general approaches:
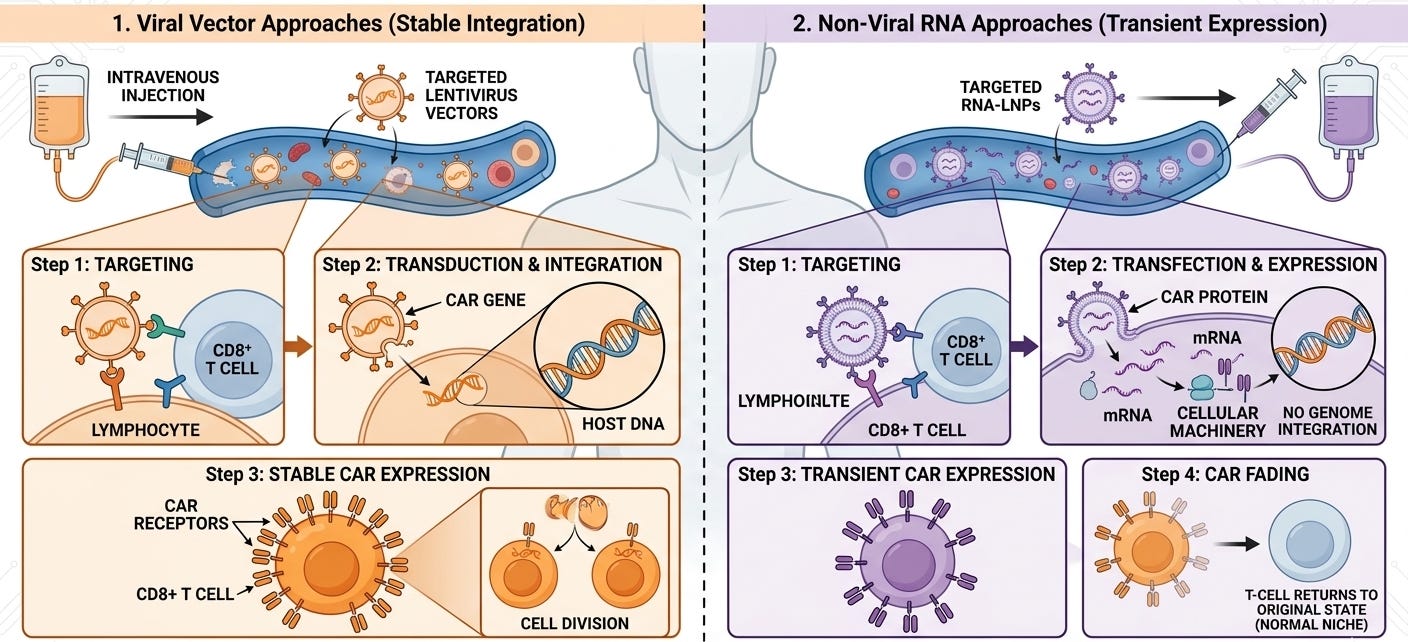
Viral Vector Approaches: Kelonia (Eli Lilly), EsoBiotech (AstraZeneca), Interius (Gilead) used hollowed-out lentiviruses that were engineered to be immune-shielded and cell-specific so they can be injected directly into the bloodstream. Their viruses used Nanobodies or scFvs to bind only to specific immune cells (like CD8+ T cells) and insert a chimeric antigen receptor (CAR) gene directly into the patient’s DNA. Since the gene is integrated into the T-cell’s DNA, the resulting CAR-T cells are longer-lasting. When the T cell divides, the CAR gene is passed on, potentially providing durable surveillance against cancer recurrence. Eli Lilly’s $7B acquisition of Kelonia (April 2026) is the current high-water mark for the field. Kelonia’s iGPS system showed 100% minimal residual disease (MRD)-negative response rate across four patients in a small Phase 1 clinical trial, with MRD-negative responses maintained through three months in the two patients with the longest follow up. This result did not require the need for toxic lymphodepleting chemotherapy.
Non-Viral RNA Approaches: Capstan (AbbVie) borrows the approach used in COVID-19 vaccines. They package mRNA inside a Lipid Nanoparticle (LNP) that has been decorated with antibodies to find T cells. The LNP delivers mRNA into the T cell. The cell’s internal machinery reads the mRNA and temporarily expresses the CAR protein on its surface. Unlike viral vectors, mRNA does not integrate into the DNA. The CAR expression eventually fades away. This could make it safer, but there isn’t data to back this up. Orna (Eli Lilly) and Orbital (Brisol-Myers Squibb) used a similar LNP approach, but loaded it with circular RNA (cRNA) instead of mRNA. Circular RNA (oRNA) has been hailed as the next generation RNA approach. While standard mRNA is a linear strand that the body breaks down quickly, circular RNA is a closed loop, which makes it more challenging for the body’s “trash-collecting” enzymes (exonucleases) to find an end to start chewing it up. Circular RNA can survive in the cell much longer than linear mRNA (one review claimed that cRNAs exhibited protein expression for 7 days compared to less than a full day for mRNA). This could allow for days of CAR expression rather than hours, without permanent DNA integration and other potential safety risks of a viral vector.
T-cell Engagers (TCE), Antibodies that Rally the Troops
T-cell engagers (TCEs) are specialized types of immunotherapies, typically bispecific antibodies, designed to bridge the gap between a patient’s immune system and a target cell (usually a cancer cell or a pathogenic B-cell). Unlike traditional antibodies that simply flag a target for destruction, a TCE act as a molecular matchmaker: it physically grabs a T-cell and pulls it into direct contact with the target cell to force an immune attack. A T-cell engager has two distinct binding arms:
The Effector Arm (anti-CD3): This arm binds to the CD3 receptor on the surface of a T-cell. This binding bypasses the normal, complex “recognition” process of the immune system and essentially “hot-wires” the T-cell into an active state.
The Target Arm (BCMA, CD19, and PSMA are the most common): This arm binds to a specific protein found on the surface of the cell you want to kill.
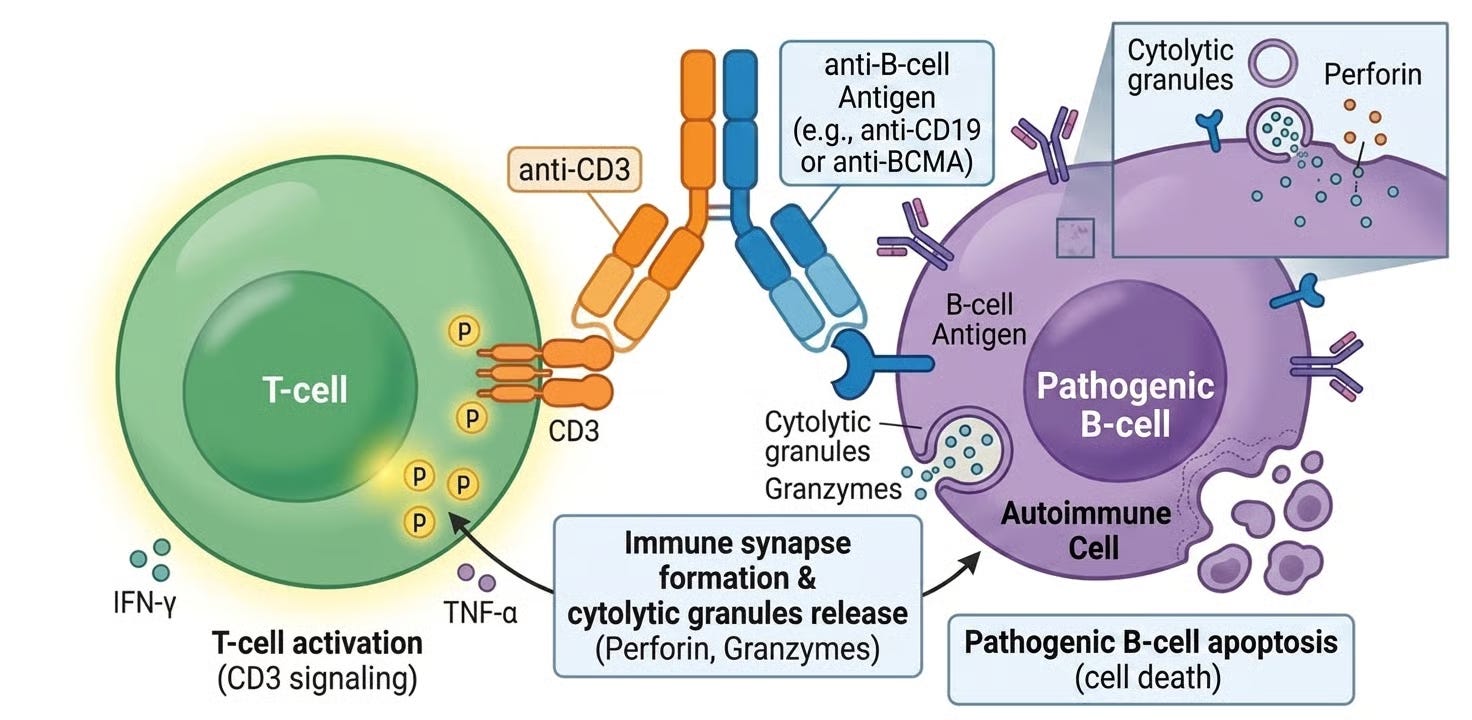
By bringing these two cells into close proximity, the TCE creates an artificial synapse. The T-cell then releases toxic enzymes (perforins and granzymes) that punch holes in the target cell, causing it to die. T-cell engagers represent a third “off-the-shelf” alternative to CAR-T therapy, an antibody that mimics the function of the cell therapy.
The development of TCE’s occurred in parallel to Dr. Michel Sadelain’s and Dr. Carl June’s development of CAR-T. The foundational spark occurred in 1985 with a landmark paper in Nature by Uwe Staerz and Michael Bevan the Scripps Research Institute in La Jolla, California. They didn’t have bispecific antibodies as we know them. Instead, they took two different monoclonal antibodies, one that targeted the T-cell receptor (TCR) and one that targeted a specific antigen, and chemically cross-linked them into a single “heteroaggregate.” They showed that these hybrid molecules could bypass the need for a T-cell to recognize a cancer cell naturally and forced a physical connection that triggered the T-cell to release its lethal payload (perforins) regardless of the cancer’s attempts to hide.
Two years after the Staerz/Bevan’s chemical heteroaggregate discovery, Mike Clark and Herman Waldmann at the University of Cambridge in the United Kingdom published the first description of a true bispecific antibody (bsAb) created through “hybrid hybridomas” (often called Quadromas). They fused two different antibody-producing cells together. The resulting cell would spit out a “mosaic” antibody that had one arm for the T-cell and one arm for the target. However, their protocol had a critical flaw: it was a manufacturing nightmare. The fused cells produced ten different combinations of antibody arms, and only one of those ten was the correct bispecific molecule. Purifying the intended bsAb was nearly impossible at the time.
In 1990, Dr. Taizo Nitta and his colleagues at the Juntendo University School of Medicine in Tokyo provided the first evidence that these “bridging molecules” could actually shrink tumors in human patients.Their findings, published in The Lancet and the Journal of Neurosurgery, remain a foundational milestone in the history of T-cell engagers (TCEs). The study focused on malignant glioma (an aggressive brain cancer), a disease chosen because it was notoriously resistant to conventional surgery and chemotherapy. They used a bispecific antibody (BsAb) created through the Quadroma (hybrid-hybridoma) method. One arm of the antibody targeted CD3 (on the T cell), and the other targeted a Glioma-Associated Antigen on the tumor cells.
The 1990 Lancet paper, titled “Preliminary trial of specific targeting therapy against malignant glioma,” reported several key findings. In a small pilot group of 10 patients, they observed significant tumor shrinkage (regression) in several cases. They proved for the first time in humans that you could kill cancer cells without the T cell needing to “naturally” recognize the tumor through the MHC system. The antibody bridge was sufficient to trigger the kill. While side effects like fever were noted, the treatment did not cause the catastrophic brain swelling that many had feared would occur when activating T cells in the confined space of the skull.
While the Lancet paper focused on the clinical outcomes, the Journal of Neurosurgery paper titled “Induction of cytotoxicity in human T cells coated with anti-glioma x anti-CD3 bispecific antibody against human glioma cells“ detailed the “how”, specifically the laboratory engineering and the cellular proof-of-concept. Instead of a simple IV drip of the TCE, they infused T-cells that were coated with a CD3 x Anti-Glioma TCE outside of the body. They harvested T cells (specifically Lymphokine-Activated Killer or LAK cells) from the patient, coated them with the bispecific antibody in a lab, and then re-infused these primed cells directly into the tumor cavity or the carotid artery.
Despite Nitta’s success, the field stalled shortly afterwards. Patients had a toxic HAMA (Human Anti-Mouse Antibody) response; their immune systems would recognize the mouse-derived antibodies as foreign and destroy them. The same fate befell Removab (catumaxomab; EpCAM x CD3 TCE). In 2009, Removab became the first TCE approved in the EU for intraperitoneal treatment of malignant ascites in adult patients with EpCAM-positive carcinomas. Being a rat-mouse hybrid, it was highly immunogenic. Patients quickly developed Human Anti-Mouse Antibodies (HAMA) and Human Anti-Rat Antibodies (HARA), which essentially neutralized the drug after a few doses, making long-term treatment difficult. By 2013-2014, marketing of Removab effectively stopped, and in 2017, the marketing authorization was officially withdrawn. The original developer, TRION Pharma, faced significant financial distress and eventually went into insolvency. The partner, Fresenius Biotech, underwent a corporate restructuring and shifted focus away from specialty oncology. Lindis Biotech (founded by the original inventor, Dr. Horst Lindhofer) spent years regaining the rights and modernizing the process. In February 2025, the European Commission granted a new EMA marketing authorization for catumaxomab (under the brand name Korjuny) for the intraperitoneal treatment of malignant ascites, marking a significant comeback for the first-ever approved TCE. Partnering with Pharmanovia, the drug is being relaunched across Europe.
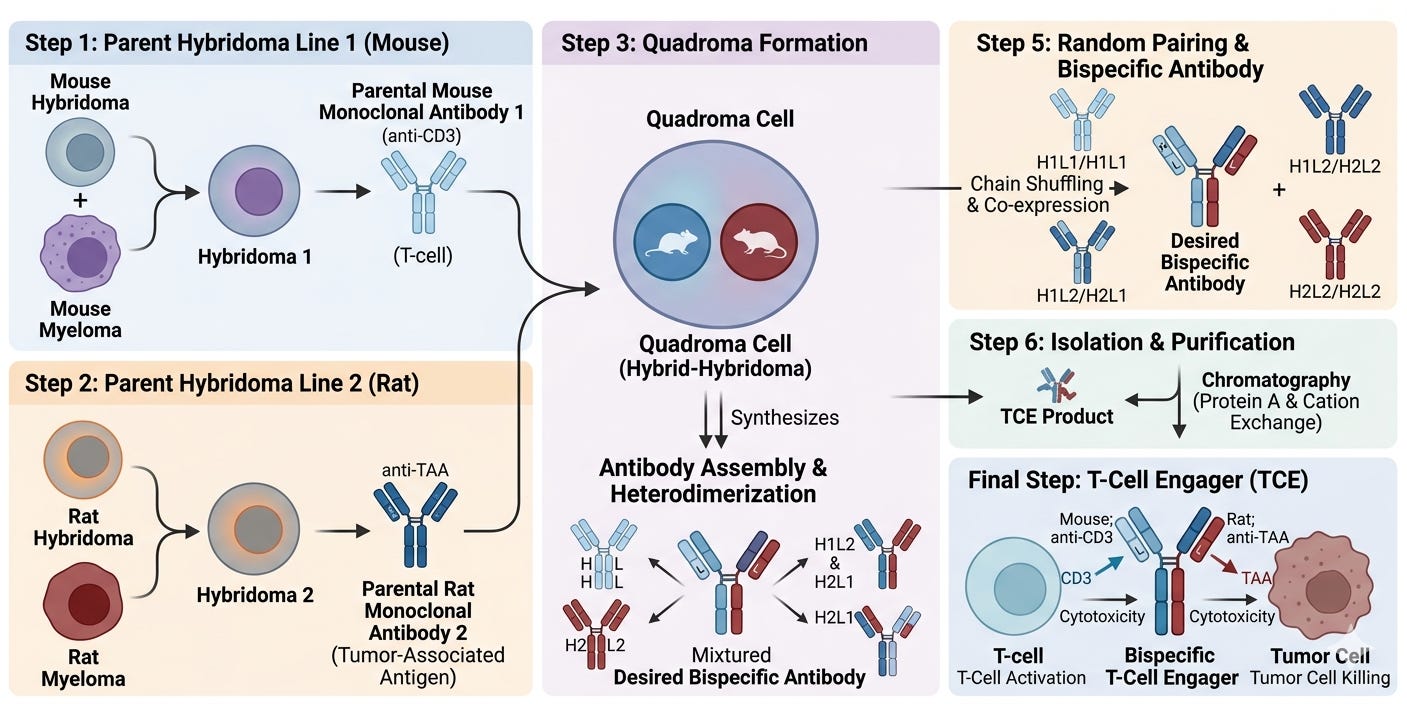
Meanwhile, Patrick Baeuerle and colleagues working at the Institute of Immunology at the University of Munich in Germany had spoun out a biotech company called Micromet in 1993. The aimed to solve the HAMA (Human Anti-Mouse Antibody) problem, though they did it through miniaturization and low dosing rather than full humanization. Micromet developed the Bispecific T-cell Engager (BiTE) platform. By stripping away the bulky Fc region of an antibody and linking only the binding arms (scFvs) with a flexible peptide chain, they created a molecule that was only 55 kDa, about 1/3 the size of a standard antibody. This smaller size was critical. It allowed the T-cell and the cancer cell to get close enough, approximately 15 nanometers, to form an immunological synapse. This helped reduce the toxic HAMA (Human Anti-Mouse Antibody) response in two ways:
Small Surface Area: By removing the Fc region of the antibody, body’s immune system had less surface area to recognize it as foreign, significantly lowering the HAMA response compared to the 1990-era drugs.
Less Drug: Since the BiTE platform was so efficient at creating an immunological synapse, it worked at incredibly low concentrations. The body can’t develop an allergic-like response (HAMA) to something that is barely present in the bloodstream. The dose was so small that it often stayed under the radar of the patient’s antibody-producing B-cells.
In 2008, Micromet published a landmark study in Science showing that incredibly low doses of their lead program blinatumomab (then called MT103) could clear tumors in patients with Non-Hodgkin Lymphoma (NHL). Before this publication, the industry was skeptical that a drug with such a short half-life and such low dosing could actually eliminate established tumors in humans. Researchers observed complete tumor regressions at a dose of just 0.005 milligrams per square meter per day. For context, that is roughly 10,000 times lower than the dose of a standard monoclonal antibody like Rituxan. Out of the patients treated at the target dose level, 100% showed a reduction of tumor cells in the blood, and several experienced complete and partial responses, even those who had failed all previous lines of chemotherapy.
Inspired by this initial positive data blinatumomab, Micromet paid to buy back the North American rights for blinatumomab from MedImmune, giving them 100% global rights to the drug. To fund their independent operations, they partnered with Sanofi for other BiTE assets, which provided non-dilutive capital and further validated the platform’s worth to external observers. In 2009, Micromet entered into a large-scale manufacturing agreement with Lonza, dispelling the skepticism that blinatumomab was un-manufacturable at scale.
They also initiated the pivotal Phase 2 BLAST trial in B-cell precursor Acute Lymphoblastic Leukemia (ALL). It was strategically designed around neutralizing Minimal Residual Disease (MRD), shifting the goalposts from treating treatment-resistant tumors to preventing the relapse. Micromet’s internal research suggested that T-cell engagers were most effective when the “T-cell to Tumor cell ratio” was in the T-cell’s favor. In late-stage patients, the sheer mass of the tumor often exhausts the T-cells. In MRD+ patients, the tumor burden is microscopic (less than 5% blasts).
Data from the Phase 2 BLAST trial were released at ASCO 2011 and ASH 2011 conferences. At the time, if a patient was MRD-positive after intensive consolidation chemotherapy, their chances of clearing that disease with more chemo were extremely low (typically <20%). Micromet showed that 16 out of 21 patients (76%–80%) with persistent or relapsed MRD became MRD-negative after just one 4-week cycle of blinatumomab. Crucially, 12 of the 16 responders were “molecularly refractory,” meaning their cancer had already seen and defeated the best chemotherapy cocktails available. Blinatumomab cleared them anyway.
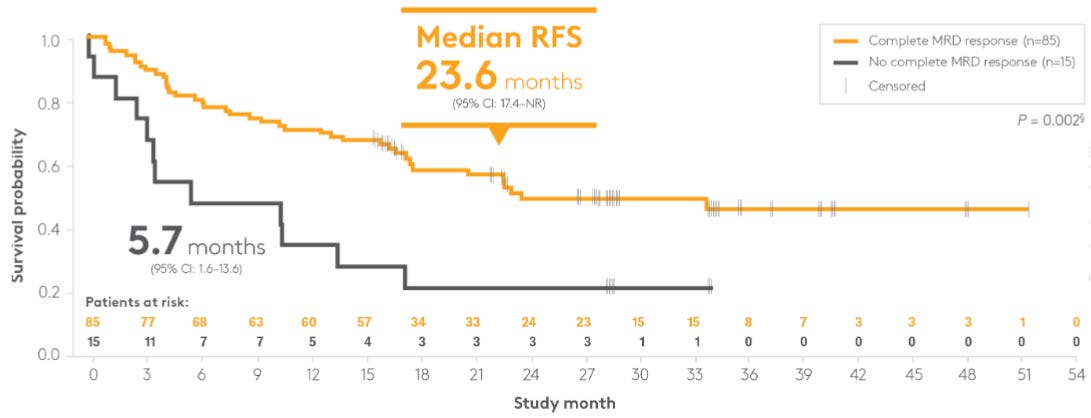
The most “buzz-worthy” part of the 2011 presentation was the early survival curve. The study reported an Relapse-Free Survival (RFS) of 78% at a median follow-up of 405 days (~13.5 months). It solidified that achieving MRD-negativity with blinatumomab was a direct predictor of long-term survival. If the drug made you MRD-negative, your risk of death or relapse dropped by nearly three-fold. Although the 2011 paper was a pilot, it set the stage for the Landmark Analysis seen in the later BLAST study, where the difference in median RFS was 23.6 months for MRD-responders vs. 5.7 months for MRD-non-responders (see Kaplan-Meier curves below).
By late 2011, Micromet had full global rights, pivotal Phase 2 data, and a proven manufacturing process. Amgen, looking to diversify beyond its aging anemia franchise (Aranesp/Epogen), moved quickly. On January 26, 2012, Amgen announced a definitive merger agreement under which to acquire Micromet for approximately $1.16 billion, a roughly 33% premium at the time. The acquisition provided three distinct layers of value:
The Lead Asset (Blinatumomab): At the time, it was in Phase 2 for acute lymphoblastic leukemia (ALL) and non-Hodgkin lymphoma (NHL). Amgen’s massive regulatory and clinical infrastructure was exactly what was needed to push it through to its 2014 approval.
The BiTE Platform: Amgen gained proprietary rights to the entire modular system, allowing them to rapidly create new TCEs by swapping out the tumor-binding arm.
The Munich Center of Excellence: Amgen chose to keep Micromet’s Munich site operational. This ensured that the original brain trust of the BiTE platform remained intact, rather than losing the institutional knowledge through a messy integration.
The Micromet acquisition is often cited as one of the cleanest exits in biotech history because the company successfully solved the three main pillars of risk: Science, Manufacturing, and Legal Rights. It was clean for Amgen as well because, in just two years in 2014, the FDA granted accelerated approval to blinatumomab (marketed as Blincyto) for relapsed/refractory (R/R) B-cell precursor ALL, making it the first FDA-approved T-cell engager (TCE).
Later on, Amgen realized that Blincyto’s real power was not in “salvage” (late-stage) therapy, but in “consolidation” (early-stage). Following the successful Phase 3 E1910 trial, the FDA expanded Blincyto’s label to include frontline consolidation for Ph- B-ALL patients, regardless of their MRD status, effectively moving the ‘molecular matchmaker’ into the earliest stages of treatment. By the late 2010s, competitors were developing TCEs that could be given as a simple weekly injection. Amgen countered by evolving the Micromet platform. Amgen researchers in Munich (the original Micromet site) successfully added a humanized Fc region to the BiTE scaffold. This increased the molecular weight and prevented rapid kidney clearance, extending the half-life from ~2 hours to several days. This paved the way for subcutaneous dosing, removing the need for the continuous infusion pump.
Amgen used the “blinatumomab blueprint” to attack other cancers, leading to the diverse 2026 portfolio you see today, including Imdelltra (tarlatamab, DLL3 x CD3 TCE). This is perhaps the most successful descendant of the Micromet acquisition. It was approved in 2024 for Small Cell Lung Cancer (SCLC), proving the BiTE platform could finally crack the code of solid tumors.
While Amgen’s Blincyto (blinatumomab, CD19 x CD3 TCE) and Imdelltra (tarlatamab, DLL3 x CD3 TCE) paved the way, the 2022–2026 period has seen a Cambrian Explosion of T-cell engagers (TCEs) from other major players. As of May 2026, the market has matured, with several TCEs moving from late-stage salvage into earlier lines of therapy and subcutaneous formats:
Roche/Genentech’s Lunsumio (mosunetuzumab, CD20 x CD3 TCE): This drug was approved for R/R Follicular Lymphoma (FL) in 2022. In December 2025, the FDA approved Lunsumio VELO, a subcutaneous version that reduced administration time from a 4-hour infusion to a one-minute injection.
Johnson & Johnson’s Tecvayli (teclistamab, BCMA x CD3 TCE): This drug is the first FDA-approved TCE targeting BCMA (2022). Its March 2026 approval in combination with Darzalex Faspro for R/R MM after just one prior line of therapy has solidified it as a standard of care bridge, similar to how Blincyto moved upstream in ALL.
Johnson & Johnson’s Talvey (talquetamab, GPRC5D x CD3 TCE): This drug is the first FDA-approved TCE targeting GPRC5D (2023). It is unique because it works for patients who have already failed BCMA-targeted therapies (like CAR-T or other TCEs).
Pfizer’s Elrexfio (elranatamab, BCMA x CD3 TCE): This drug was approved for R/R multiple myeloma (MM) in 2023. In 2025-2026, it gained traction due to its fixed-dose subcutaneous schedule that moves to every-four-week dosing for responders, potentially increasing convenience for patients.
Regeneron’s Lynozyfic (linvoseltamab, BCMA x CD3 TCE): This drug was recently approved in 2025 for heavily pre-treated MM. Regeneron is currently betting big on MRD-negativity (Phase 3 data due in 2028) to potentially challenge J&J’s dominance in frontline settings.
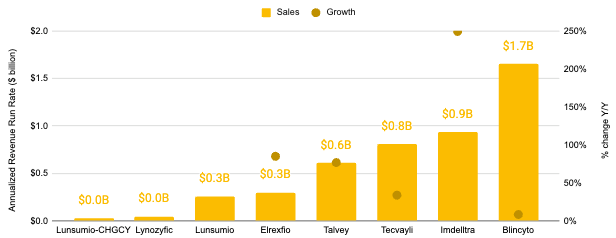
While Amgen dominates CD19 and DLL3, J&J and Pfizer have competed for the BCMA space, with J&J carving out a niche in GPRC5D to handle post-BCMA relapses. As of 4Q 2025, there are at least 87 clinical-stage TCE programs. T-cell engagers (TCEs) constitute a blockbuster therapeutic modality, representing an estimated aggregate revenue run rate of more than $4.6 billon as of 4Q 2025 according to quarterly press releases associated with Blincyto, Imdelltra, Tecvayli, Talvey, Elrexfio, Lunsumio, and Lynozyfic* (see below)
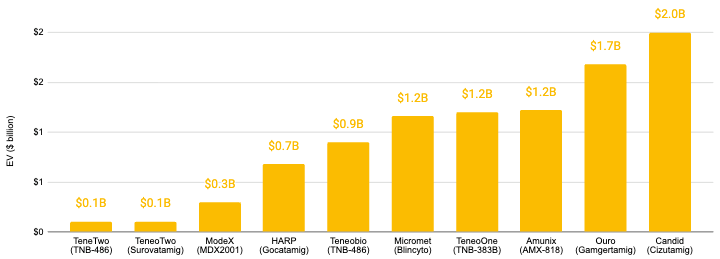
The acquisition landscape for T-cell engagers (TCEs) are also quite healthy, representing more than $9 billion in aggregate enterprise value (EV). We recently covered the largest TCE acquisition to date, UCB’s acquisition of Candid Therapeutics for $2 billion in upfront payments and up to $200 million in potential future milestone payments ($2.2 billion total).
Conclusion
The journey from the first personalized CAR-T infusions to the modern era of allogeneic cells, in vivo engineering, and T-cell engagers represents a significant evolution in how we think about living drugs. We are moving away from the bespoke model of CAR-T, where every treatment is handcrafted for a single patient, and toward an “off-the-shelf“ future that is faster, cheaper, and more accessible. The milestones we’ve witnessed are dismantling of the barriers that have historically kept these treatments confined to elite academic centers:
Allogeneic CAR-T is proving that we can use healthy donor cells to treat patients in days rather than weeks, as seen with early clinical cema-cel’s success in outpatient management.
In vivo CAR-T promises to turn the human body itself into a manufacturing plant, potentially removing the need for toxic lymphodepleting chemotherapy.
T-cell Engagers (TCEs) like Blincyto have brought the power of T-cell recruitment into the realm of half-life extended antibodies that require only weekly subcutaneous injections.

As we look toward Part 3, the CAR-T’s horizon expands even further. The same tools that were built to hunt down aggressive liquid tumors are now being re-tooled to reset the immune system itself. Stay tuned for Part 3 of this three-part Frontiers in Medicine series on CAR-T, where we explore how CAR-T is moving beyond oncology to tackle the challenge of autoimmune diseases, potentially offering a multi-year reset for patients with Lupus, Myasthenia Gravis, and beyond.
If you missed Part 1 of this three-part Frontiers in Medicine series on CAR-T, check it out here. It discussed the Immunology Civil War, the discovery of T-cells, and their rebirth as medicines.
To contact us, please send us an email at biotechreadout@gmail.com
Disclaimers
Investigational Status Disclaimer
The therapeutic candidates discussed in this newsletter are currently in clinical development and have not been approved for commercial sale by the U.S. Food and Drug Administration (FDA), the European Medicines Agency (EMA), or other global regulatory authorities. Their safety and efficacy have not been established. References to pipeline products and ongoing clinical trials involve significant risks and uncertainties. Statements regarding the potential safety, potency, or efficacy of investigational drugs reflect current hypotheses and are not a guarantee of future performance or regulatory clearance. The outcome of clinical trials is inherently unpredictable, and clinical results from earlier stages may not be predictive of results in later, larger-scale trials.
No Medical Advice Disclaimer
This newsletter is for informational and educational purposes only. The content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or another qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read in this publication.
No Patient-Provider Relationship Disclaimer
The information provided in this newsletter is for educational and analytical purposes only. Receipt of this information, or any interaction with this content, does not create a physician-patient, pharmacist-patient, or any other professional-provider relationship between you and the authors or publishers. This newsletter should not be used as a substitute for a personal consultation with a qualified healthcare professional.
Forward-Looking Statements Disclaimer
This newsletter contains “forward-looking statements” regarding future events, including clinical trial timing, regulatory milestones, and projected market performance. These statements are based on current expectations and assumptions that are subject to significant risks and uncertainties. Actual results may differ materially from those expressed or implied. We undertake no obligation to update these statements as a result of new information or future developments.
Third-Party Links & Content Disclaimer
This newsletter contains links to third-party websites, including clinical trial registries and corporate presentations. Biotech Readout does not endorse, guarantee, or assume responsibility for the accuracy or reliability of any information offered by third-party providers.
Errors and Omissions Disclaimer
While we strive for technical accuracy, the information in this newsletter is provided on an “as is” basis with no guarantees of completeness, accuracy, or timeliness. Biotech Readout assumes no liability for any errors or omissions in the content of this publication.
Non-Endorsement Disclaimer
Any reference to specific commercial products, processes, or services by trade name, trademark, or manufacturer does not constitute or imply an endorsement or recommendation by the author. All trademarks are the property of their respective owners.
No Investment Advice Disclaimer
This newsletter is for informational purposes only and does not constitute financial, investment, or legal advice. The author is not a registered investment advisor. You should consult with a professional financial advisor before making any investment decisions. The biotechnology sector is highly volatile; past performance is not indicative of future results.
Conflict of Interest Disclaimer
The author of this newsletter maintains a position of independence. At the time of publication, the author holds no direct financial interest, equity, or options in any of the companies mentioned in this report. No compensation has been received from any third party to feature or analyze specific therapeutic candidates or corporate entities.