
Ipsen to Acquire Kartos
Unleashing apoptotic p53 on myelofibrosis

Disclaimer: This newsletter is for educational and informational purposes only and does not constitute medical, investment, or financial advice, nor does it establish a provider-patient relationship. Content may include forward-looking statements and discussions of investigational therapeutic candidates that are not FDA/EMA approved; their safety and efficacy remain unestablished and clinical outcomes are unpredictable. While we strive for accuracy, all information is provided as is without guarantees. As of the date of publication, the author holds no direct equity positions in the specific companies mentioned in this issue nor receives third-party compensation for this coverage. Please find a complete version of our disclaimers at the bottom of this article and on our About page.

Introduction
On June 29, 2026, Ipsen announced a definitive agreement to acquire Kartos Therapeutics for $450 million upfront at closing and up to $1.3 billion of regulatory and sales-based milestones, for a total deal value of up to $1.75 billion. This acquisition centers on navtemadlin, an oral MDM2 inhibitor that is currently being evaluated in a Phase 3 myelofibrosis trial. This is the second acquisition in the myelofibrosis space in about two months, with the first being Eli Lilly’s $2.3 billion purchase of Ajax Therapeutics.

In this article, we explore the recent interest in myelofibrosis acquisitions and unravel the novel mechanism behind Kartos’ lead molecule.
The Duality of Myelofibrosis
In Roman mythology, Janus is the god of duality. He is uniquely depicted with two faces looking in opposite directions, a representation known as Janus Bifrons (Janus Two-Faced). The two heads symbolize fundamental dualities of the human experience and the natural universe.

Myelofibrosis has a duality of its own: it is both big (by sales) and small (by the number of R&D candidates). As of the 1Q 2026, FDA-approved myelofibrosis medicines account for an almost $6.3 billion annualized revenue run rate, placing the indication comfortably in the blockbuster zone. Nevertheless, clinical-stage R&D pipeline is rather sparse. Ironically, the duality of myelofibrosis’ big market and small competition has emerged due to biotech/pharma crowding around inhibitors of Janus kinase (JAK), a protein named after Roman god Janus.
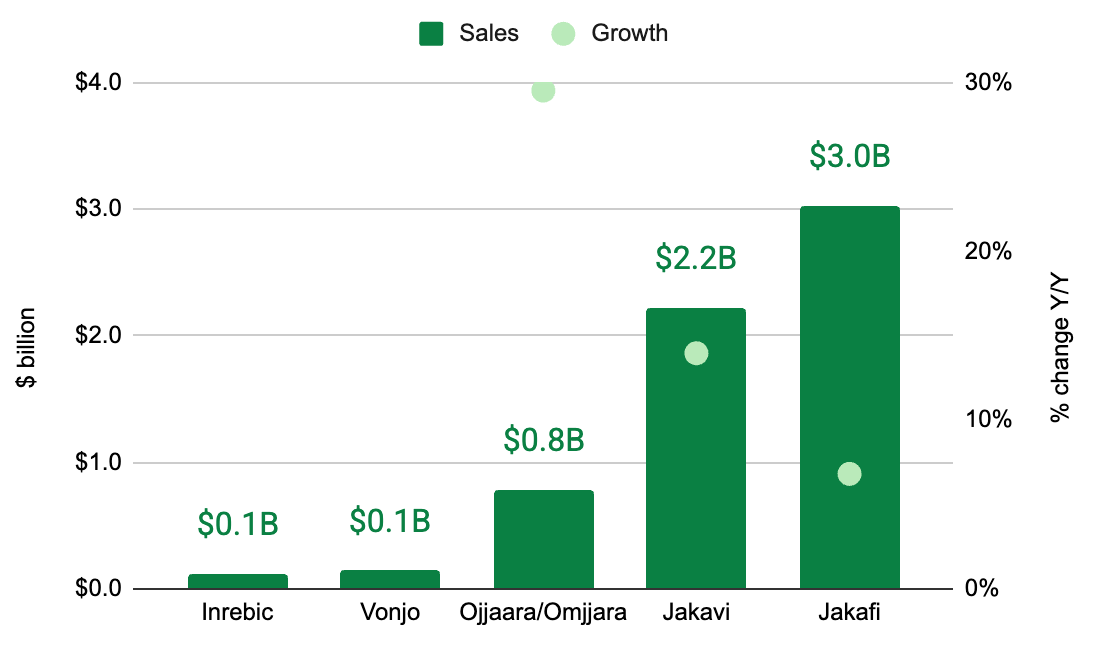
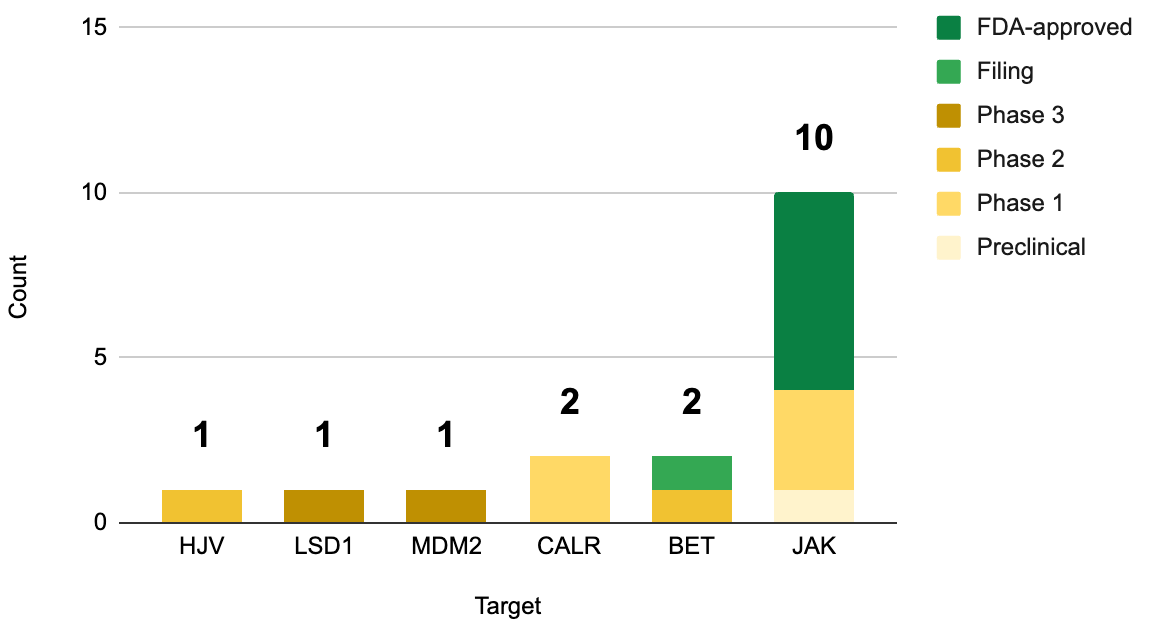
As of June 2026, all FDA-approved myelofibrosis drugs target JAK. To be clear, they each have their nuances. They span two generations, each with unique safety and efficacy profiles, but all target JAK nonetheless (we covered that history here). Of course the relentless the pursuit of JAK inhibitors that isn’t surprising, given that JAK mutations are the most common genetic drivers of primary myelofibrosis (50-60% of cases), followed by CALR mutations (25-35% of cases; 2 mutant CALR inhibitors in development) and MPL mutations (remaining 5-10% of cases; no drugs directly targeting this). Kartos’ navtemadlin stands alone as the first and only myelofibrosis drug that targets MDM2.
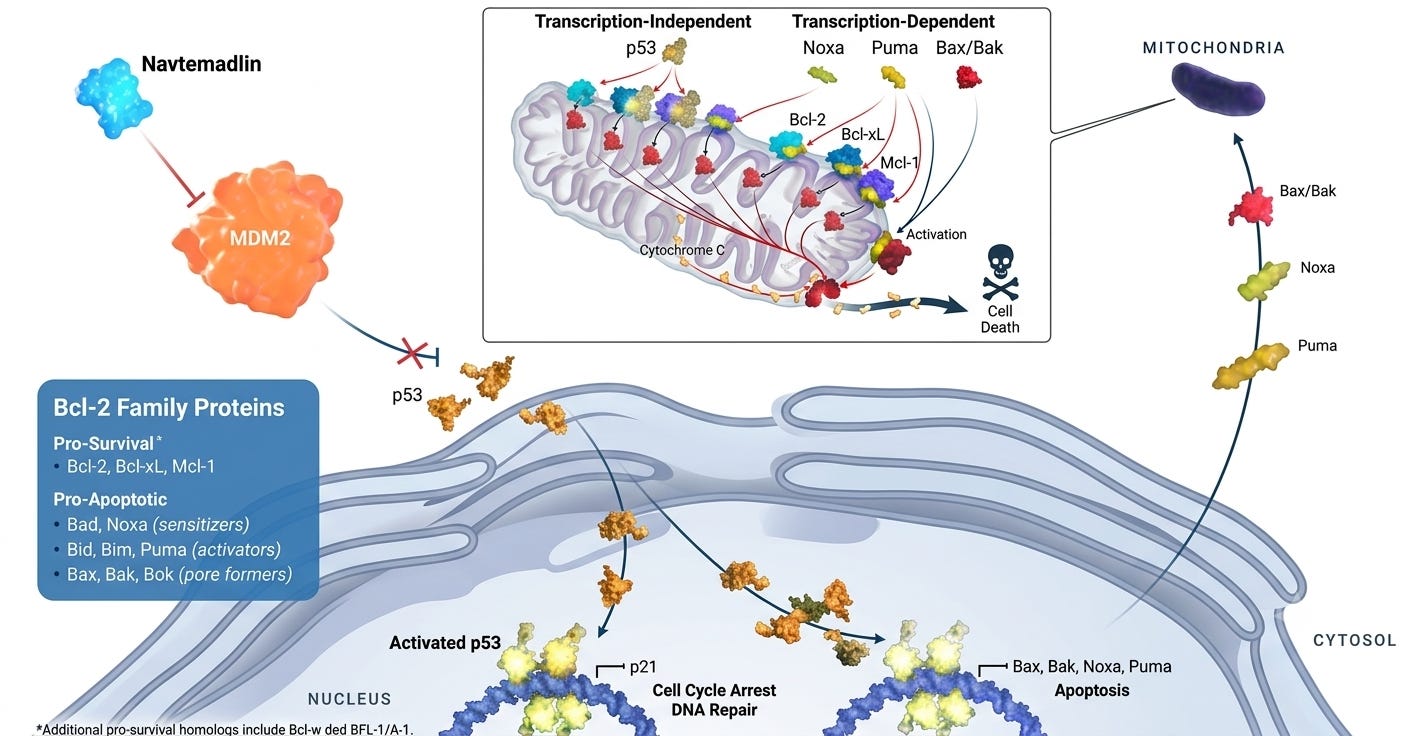
Programming Cancer Cell Death
The central thesis of Kartos’ navtemadlin centers on harnessing the therapeutic potential of the p53 pathway. In many cancers, the crucial tumor-suppressor protein p53 remains un-mutated (wild type), but is functionally inactivated by the overexpression of its negative regulator, MDM2. By blocking the MDM2-p53 interaction, Kartos’ candidate is designed to force the reactivation of p53 to drive apoptosis in cancer cells. In other words, navtemadlin a de-represses of programmed cell death. If it’s confusing to you, don’t be alarmed. The legendary scientists who discovered p53 were just as baffled.
The path to uncovering the role of p53 in oncology is one of the most famous plot twists in biomedical history. When p53 was first discovered in 1979 by several independent groups (including David Lane, Arnold Levine, and Lloyd Old), it was completely mischaracterized. Researchers isolated p53 because it physically bound to the SV40 Large T-antigen, a viral protein known to transform normal cells into cancerous ones. Since high levels of p53 were found in tumor cells but were nearly undetectable in healthy cells, scientists reasoned that p53 was an oncogene, a gene that accelerated tumor growth. This theory was accidentally formulated when early labs cloned p53 DNA and showed it could transform healthy cells in vitro. What they didn’t realize was that they had unknowingly cloned a mutated isoform of p53 DNA rather than the normal, wild-type version.
In 1989, Arnold Levine’s lab and Bert Vogelstein’s team discovered that wild-type p53 actually inhibited cell transformation. Vogelstein found that the TP53 gene was frequently deleted or mutated in human colorectal cancers. The scientific community realized they had it backward: p53 was a tumor suppressor gene (slows tumor growth), not an oncogene (accelerates tumor growth). When functioning correctly, wild-type p53 acts as a transcription factor activated by cellular stress (like DNA damage or low oxygen). It forces the cell into cell-cycle arrest to repair the damage or triggers apoptosis if the damage is catastrophic. If p53 is mutated or inactivated, cells can accumulate genetic mutations unchecked, paving the way to malignancy.
How does this relate to myelofibrosis? For years, the molecular focus remained largely fixed on its three genetic driver mutations (JAK, CALR, MPL). However, next-generation sequencing and longitudinal tracking revealed that p53 plays a crucial, distinct role in disease architecture and progression. While p53 mutations are relatively rare in the early, chronic phases of primary or secondary myelofibrosis (occurring in only 3-5% of patients), their emergence is heavily tied to blast-phase progression (transformation into secondary Acute Myeloid Leukemia, or sAML). Once a myelofibrosis clone acquires a TP53 mutation, the cells rapidly accrue massive chromosomal structural variations and deletions.
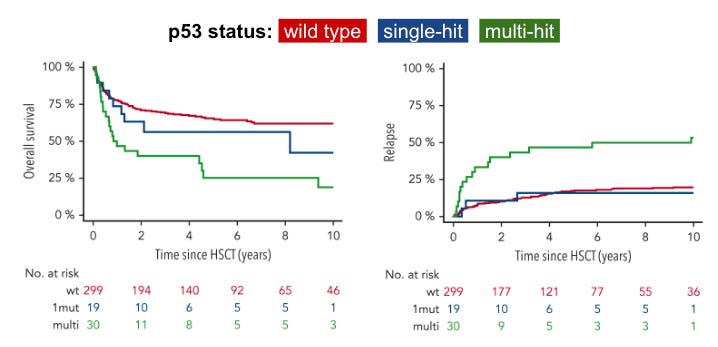
The status of p53 mutations in myelofibrosis significantly impacts overall survival and relapse incidence, according to retrospective cohort data from a 349-patient study of patients with myelofibrosis undergoing an allogeneic hematopoietic stem cell transplantation (HSCT) published in Blood. Patients with a mutation in only one allele, while the other remains wild-type, often experience a clinical course similar to non-mutated patients. When a patient suffers a multi-hit mutation event, such as losing the second allele through a deletion (like 17p deletion) or a second mutation, their prognosis drops dramatically. Multi-hit TP53 status is associated with about half the overall survival after 6 years (only 25% of multi-hit survived versus 56% single-hit, P< 0.001) and is associated with a 42% higher risk of disease recurrence compared to patients with single-hit TP53 status (P=0.03), following a haematopoietic stem cell transplant (HSCT).
Crucially, even when the TP53 gene is not mutated in myelofibrosis, the pathway is frequently hijacked. In many high-risk MF patients, the wild-type p53 protein is functionally suppressed because the cell overexpresses MDM2, a principal negative regulator that tags p53 for degradation. MDM2 has “high expression” in 71% of CD34+ hematopoietic progenitor cells from patients with myelofibrosis compared to 34% in healthy people (see graph below). In fact, Nakatake et al. Oncogene (2012) suggests that JAK2V617F driver mutations may cause MDM2 overexpression via the abnormal accumulation of the La autoantigen (a protein that significantly boosts MDM2 protein levels by binding & stabilizing MDM2 mRNA). This overabundance of MDM2 forces the continuous degradation of p53, preventing apoptosis even when the cell undergoes DNA damage. By either downregulating the La protein or treating the cells with an MDM2 antagonist (using Nutlin-3 as a tool compound), the team successfully rescued the p53 response to DNA damage and restored normal cytokine dependence to JAK2V617F mutant cells, curbing their unchecked growth.
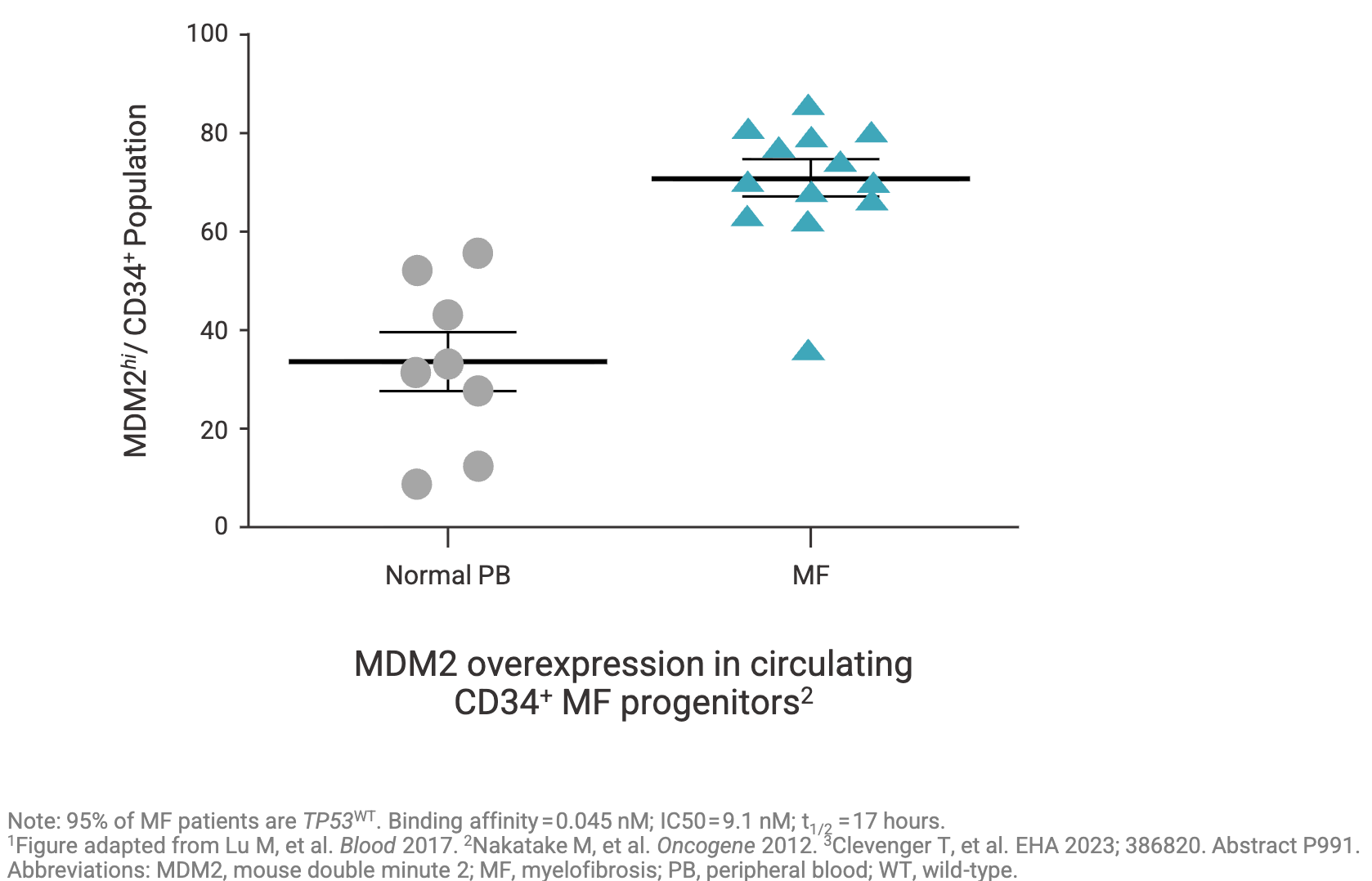
This biological interplay is precisely where Kartos’ lead program steps into the spotlight. Navtemadlin is designed to restore apoptotic p53 signaling by blocking MDM2-mediated degradation. In a therapeutic landscape heavily saturated with successive generations of JAK inhibitors, Kartos’ investigational candidate stands out as a mechanically distinct approach under evaluation for patients who have exhausted standard options.
Spinning Out
The founding of Kartos Therapeutics is a classic story of biotech spin-outs, where a highly promising molecule developed by a pharma giant was given a dedicated, laser-focused home to reach its clinical potential. Its scientific foundation started inside the research engines of Amgen, where navtemadlin was originally developed. While the molecule (then known as AMG 232) showed immense structural promise, pharmaceutical giants often have to ruthlessly prioritize their vast pipelines. Recognizing the asset’s potential but needing a dedicated team to push it through complex clinical development, Amgen looked for a strategic way to externalize the program.
In 2018, Kartos Therapeutics was officially launched to breathe new life into the asset, establishing its operations with dual headquarters in Redwood City, California, and Bellevue, Washington. The company was co-founded by industry veterans, most notably Jesse McGreivy, who stepped in as Chief Medical Officer (CMO) and Chief Executive Officer (CEO). McGreivy brought immense credibility to the venture, having previously served as a CMO at Acerta Pharma, where he played a pivotal role in the clinical development of the blockbuster blood cancer drug Calquence (acalabrutinib) before its $7 billion acquisition by AstraZeneca in 2016. Upon launch, Kartos secured an exclusive worldwide licensing agreement with Amgen for the MDM2 inhibitor, renaming the drug candidate KRT-232 (later given the generic name navtemadlin). This molecule became the sole cornerstone of the company’s pipeline. The company secured robust financial backing from premier life science institutional investors, including SR One and OrbiMed Advisors, and got straight to work.
Shortly after licensing the molecule, Kartos launched a broad clinical screening strategy. Since MDM2 inhibition restores wild-type p53 to trigger apoptosis, the company initiated parallel Phase 1 and Phase 2 trials across several oncology indications. Navtemadlin was clinically tested in multiple p53 wild-type malignancies, including acute myeloid leukemia (AML), polycythemia vera (PV), and Merkel cell carcinoma (MCC). While solid tumor cohorts encountered the traditional dose-limiting toxicities historic to the MDM2 class (primarily thrombocytopenia and gastrointestinal distress), a more favorable therapeutic window emerged in myelofibrosis.
These efforts culminated in the open label Phase 3 BOREAS trial, which represents a landmark attempt to introduce a non-JAK-inhibitor mechanism to a patient population who have relapsed or become refractory to frontline JAK inhibitor therapies. The study enrolled 183 patients with confirmed wild-type p53 primary or secondary myelofibrosis. They wanted to address the true unmet need, so approximately 65% of their recruited patients were refractory to previous JAK inhibitors, 30% presented with severe baseline thrombocytopenia (platelets < 100,000 / μL), and over 60% carried high molecular risk mutations. Topline clinical data and correlative biomarker data were first presented at the 66th American Society of Hematology (ASH) Annual Meeting in December 2024.
Navtemadlin tripled the percent of patients who achieved a ≥35% reduction in spleen volume compared to best available therapy at week 24 (SVR35: 15% versus 5%), although it narrowly missed statistical significance (P=0.0815) due to adjustments in the trial’s accrual targets. It also doubled the percent of patients who experienced a ≥50% reduction in disease-related constitutional symptoms compared to best available therapy (TSS50 24% versus 12%, P=0.05). While the primary endpoint did not achieve traditional statistical significance in this specific cohort, investigators noted that the observed data trends provided a rationale for further clinical evaluation of navtemadlin’s potential impact on tumor bulk and constitutional symptoms.
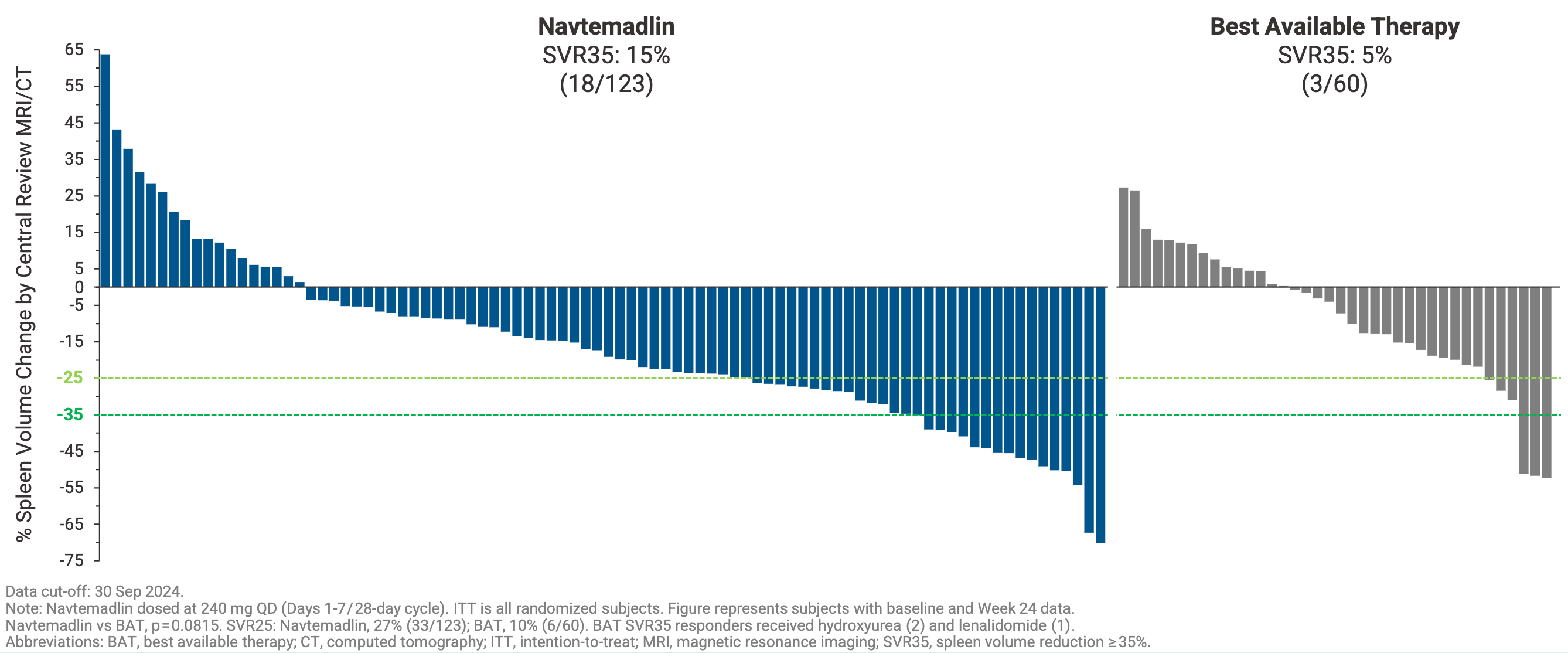
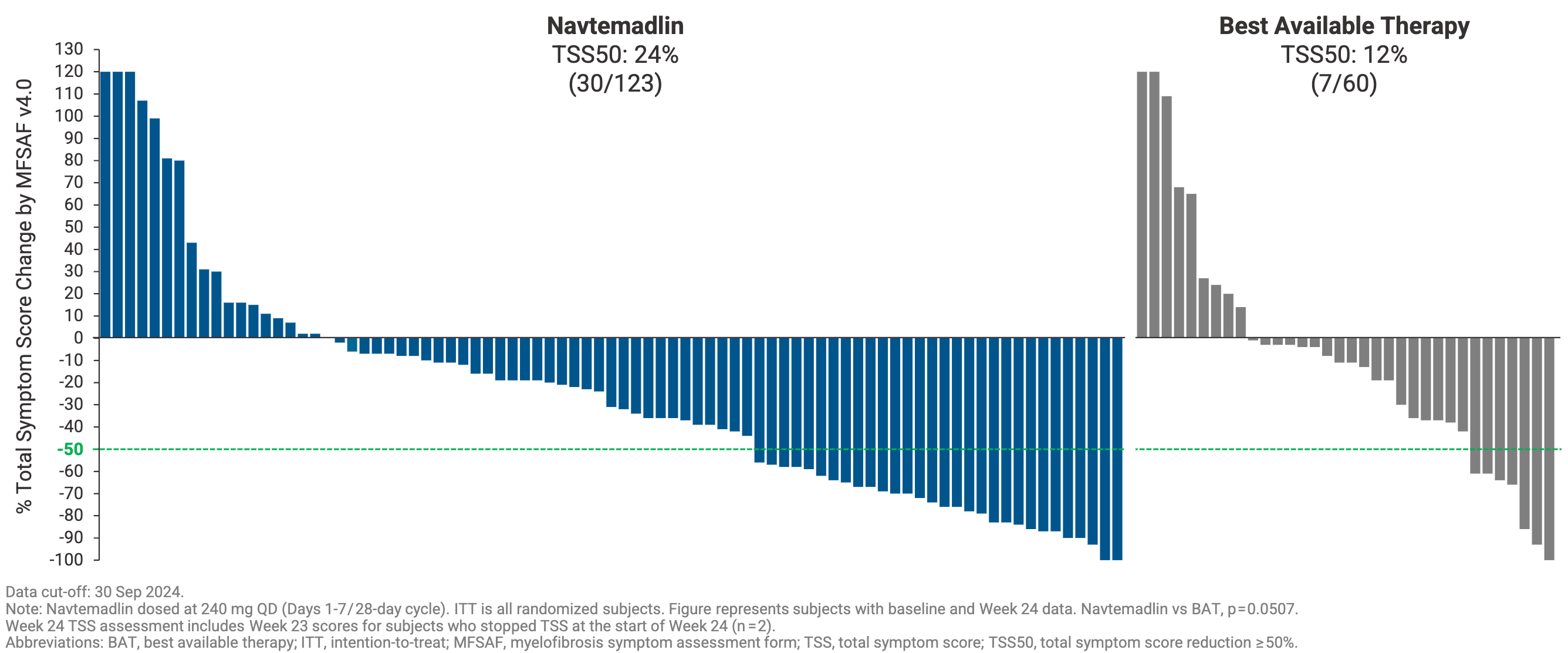
Where BOREAS truly generated excitement in the hematology community was in its exploratory, correlative biomarker readouts. Unlike JAK inhibitors, which primarily mask symptoms, navtemadlin demonstrated anti-clonal activity based on three orthogonal readouts:
CD34+ Cell Clearance: Circulating CD34+ cells, the hallmark malignant driving cells of myelofibrosis, dropped -70% at Week 24 in the navtemadlin arm compared to just -38% in the BAT arm.
Variant Allele Frequency (VAF): Navtemadlin doubled the molecular response rate, achieving a ≥50% reduction in the underlying driver gene VAF (such as JAK2 or CALR) in 21% of patients versus 12% for BAT.
Bone Marrow Fibrosis Reversal: At 24 weeks, nearly half of the evaluable navtemadlin patients (48%) achieved a documented reduction of at least 1 grade in bone marrow fibrosis, compared to just 24% in the control wing.
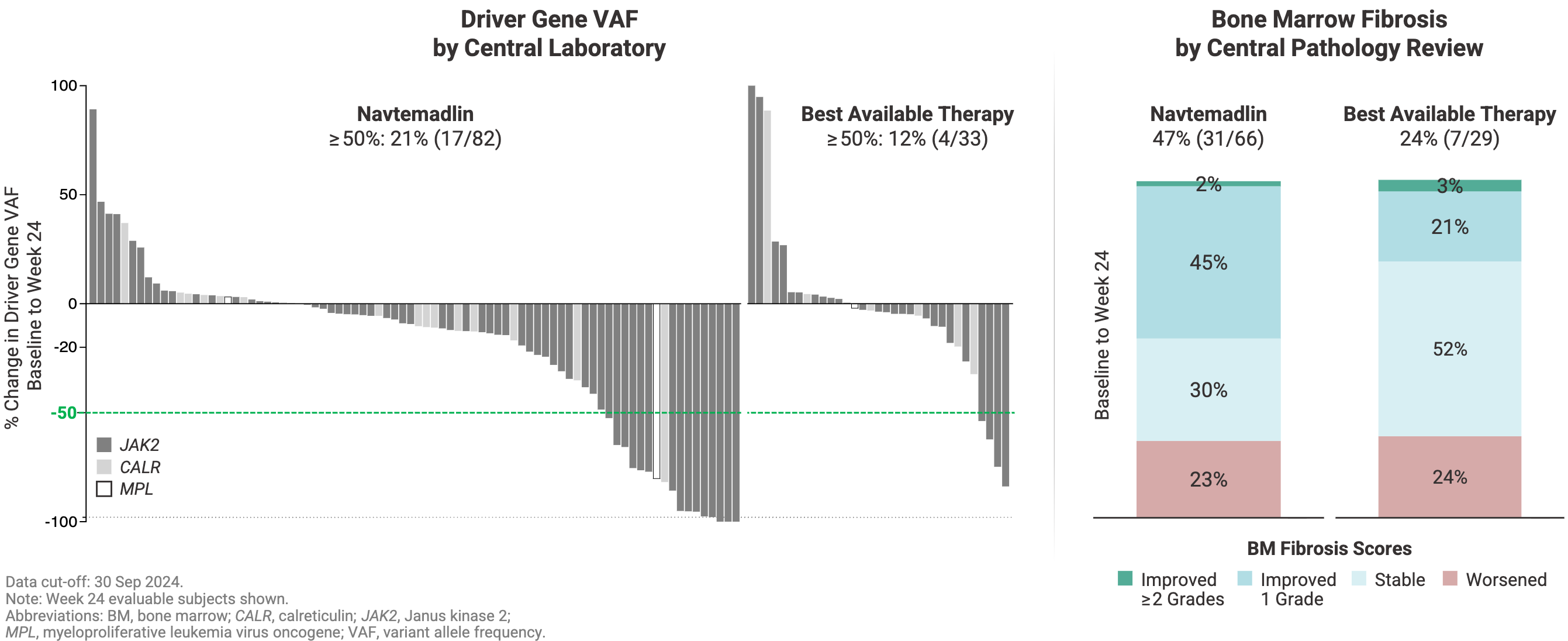
The adverse event profile yielded the expected on-target effects of the MDM2 inhibitor class, characterized by hematologic suppression and gastrointestinal distress. The enrollment of the BOREAS trial was eventually halted prior to its original total accrual target. This was a deliberate corporate pivot. The Kartos team believed that they had enough evidence from BOREAS to convince them that navtemadlin possessed robust monotherapy activity and genuine disease-modifying capability. With proof-of-concept secured, Kartos chose to push navtemadlin into a pivotal Phase 3 POIESIS trial, testing it as an add-on combination therapy to rescue suboptimal frontline Jakafi responders before their disease progresses to a completely relapsed/refractory state. It’s a much needed addition given that 60-70% of myelofibrosis patients treated with the frontline JAK inhibitor Jakafi fail to hit optimal clinical responses (defined as ≥35% spleen volume reduction and ≥50% symptom improvement). Ipsen plans to report topline data from POIESIS are expected in 2027.
Conclusion
For years, the myelofibrosis field remained tethered to JAK inhibition, a class that masterfully treats symptoms but is prone to resistance in a majority of patients. By charting a course as the MDM2 lone wolf, Kartos’ navtemadlin forged a new therapeutic approach. In doing so, it taps into a legacy of p53 biology nearly fifty years in the making. As Ipsen prepares to steer this molecule through the global expanse of the Phase 3 POIESIS trial, we will soon see whether the p53 pathway can be safely harnessed to alter the trajectory of myelofibrosis.
Love biotech? Check out Biotech Readout’s full content library, or navigate directly to a segment that interests you:
Frontiers in Medicine: Exploring the frontiers of our understanding and treatments for disease.
Medical History: Recovering forgotten relics in the history of medicine.
Acquisitions: Exploring the innovation behind acquired companies.
Weekly Readout: A digest of new clinical data from the past week.

To contact us, please send us an email at biotechreadout@gmail.com
Disclaimers
Investigational Status Disclaimer
The therapeutic candidates discussed in this newsletter are currently in clinical development and have not been approved for commercial sale by the U.S. Food and Drug Administration (FDA), the European Medicines Agency (EMA), or other global regulatory authorities. Their safety and efficacy have not been established. References to pipeline products and ongoing clinical trials involve significant risks and uncertainties. Statements regarding the potential safety, potency, or efficacy of investigational drugs reflect current hypotheses and are not a guarantee of future performance or regulatory clearance. The outcome of clinical trials is inherently unpredictable, and clinical results from earlier stages may not be predictive of results in later, larger-scale trials.
No Medical Advice Disclaimer
This newsletter is for informational and educational purposes only. The content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or another qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read in this publication.
No Patient-Provider Relationship Disclaimer
The information provided in this newsletter is for educational and analytical purposes only. Receipt of this information, or any interaction with this content, does not create a physician-patient, pharmacist-patient, or any other professional-provider relationship between you and the authors or publishers. This newsletter should not be used as a substitute for a personal consultation with a qualified healthcare professional.
Forward-Looking Statements Disclaimer
This newsletter contains “forward-looking statements” regarding future events, including clinical trial timing, regulatory milestones, and projected market performance. These statements are based on current expectations and assumptions that are subject to significant risks and uncertainties. Actual results may differ materially from those expressed or implied. We undertake no obligation to update these statements as a result of new information or future developments.
Third-Party Links & Content Disclaimer
This newsletter contains links to third-party websites, including clinical trial registries and corporate presentations. Biotech Readout does not endorse, guarantee, or assume responsibility for the accuracy or reliability of any information offered by third-party providers.
Errors and Omissions Disclaimer
While we strive for technical accuracy, the information in this newsletter is provided on an “as is” basis with no guarantees of completeness, accuracy, or timeliness. Biotech Readout assumes no liability for any errors or omissions in the content of this publication.
Non-Endorsement Disclaimer
Any reference to specific commercial products, processes, or services by trade name, trademark, or manufacturer does not constitute or imply an endorsement or recommendation by the author. All trademarks are the property of their respective owners.
No Investment Advice Disclaimer
This newsletter is for informational purposes only and does not constitute financial, investment, or legal advice. The author is not a registered investment advisor. You should consult with a professional financial advisor before making any investment decisions. The biotechnology sector is highly volatile; past performance is not indicative of future results.
Conflict of Interest Disclaimer
The author of this newsletter maintains a position of independence. At the time of publication, the author holds no direct financial interest, equity, or options in any of the companies mentioned in this report. No compensation has been received from any third party to feature or analyze specific therapeutic candidates or corporate entities.