
Eli Lilly to Acquire Ajax
A differentiated JAK play

Disclaimer: This newsletter is for educational and informational purposes only and does not constitute medical, investment, or financial advice, nor does it establish a provider-patient relationship. Content may include forward-looking statements and discussions of investigational therapeutic candidates that are not FDA/EMA approved; their safety and efficacy remain unestablished and clinical outcomes are unpredictable. While we strive for accuracy, all information is provided as is without guarantees. This newsletter is independent, and the author holds no financial positions in the companies mentioned nor receives third-party compensation for this coverage. Revenue run rates are estimates based on the author’s synthesis of publicly available 10-K/10-Q filings and may not reflect GAAP-certified annual totals. Please find a complete version of our disclaimers at the bottom of this article and on our About page.

Introduction
On April 27, 2026, Eli Lilly and Company announced a definitive agreement to acquire Ajax Therapeutics for up to $2.3 billion in cash, inclusive of an upfront payment and subsequent payments upon the achievement of certain clinical and regulatory milestones. This acquisition centers on Ajax’s lead program AJ1-11095, a small molecule JAK2 inhibitor currently in a Phase 1 clinical trial called AJX-101 (NCT06343805). Eli Lilly emphasized that the primary value-add for AJ1-11095 is its ability to selectively bind and inhibitor the Type II conformation of JAK2, whereas other JAK inhibitors typically bind its Type I conformation. This could potentially provide two points of differentiation:
Provide a novel treatment option for those patients who become resistant to Type I JAK2 inhibitors. This is currently being evaluated in the Phase 1 AJX-101 trial, which has recruited patients with myelofibrosis who have previously been treated with a Type I JAK2 inhibitor.
Deliver deeper and more durable efficacy than existing JAK2 inhibitors. This hypothesis isn’t yet being evaluated in a clinical trial.
Myeloproliferative neoplasms (MPNs) like myelofibrosis (MF) and polycythemia vera (PV) already constitute a blockbuster indication space, representing an aggregate revenue run rate of more than $6.6 billion per year and growing 13% year over year as of 4Q 2025 according to quarterly press releases associated with Jakafi/Jakavi, Ojjaara/Omjjara, Vonjo, and Inrebic*. Despite this, the competitive field is relatively sparse compared to other indication spaces with 4 approved medicines and 9 in clinical development.
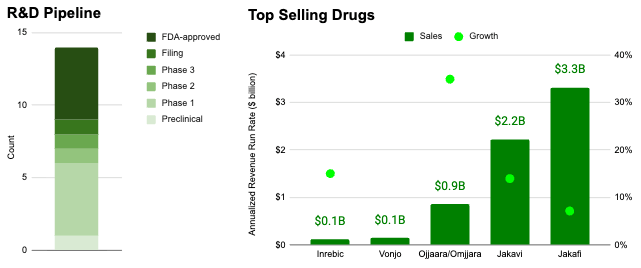
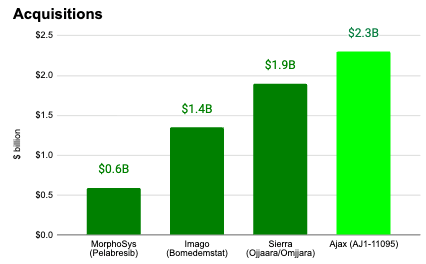
If executed, the proposed Ajax acquisition would constitute the largest acquisition in the MF space to date ($2.3B versus $1.9B for Sierra Oncology, $1.4B for Imago Bioscience, and $600M for MorphoSys).
Here, we dive into the history of myelofibrosis, polycythemia vera, and JAK inhibitors and also explore how Ajax Therapeutics’ lead program could advance the cutting edge of treatment.
Double, Double Toil and Trouble
The discovery and clinical classification of myelofibrosis (MF) and polycythemia vera (PV) span over 150 years, evolving from isolated case observations in the 19th century to the unified molecular understanding we have today. Before they were understood as related blood cancers, PV and MF were described as distinct, puzzling medical anomalies.
The first definitive description of what we now call primary myelofibrosis is credited to the German physician Dr. Gustav Heuck. In 1879, he detailed two cases of leukemia that were atypical because they featured significant bone marrow scarring (fibrosis) and massive enlargement of the spleen and liver (extramedullary hematopoiesis). The disease manifests in a debilitating clinical profile that includes splenomegaly (massive splenic enlargement due to extramedullary hematopoiesis), constitutional symptoms (severe fatigue, night sweats, and bone pain), cytopenias (progressive anemia and compromised marrow function), and reduced survival.
The French physician Dr. Louis Henri Vaquez provided the first description of PV. He observed a 40-year-old male patient with chronic cyanosis (bluish skin), distended veins, and a massive red blood cell count. In 1903, the renowned Dr. William Osler published a series of similar cases, further cementing the condition in medical literature, which was sometimes referred to as “Vaquez-Osler disease.”
For decades, these conditions were treated as separate diseases. The turning point came in 1951, when Dr. William Dameshek, a founding editor of the journal Blood, published a seminal editorial. He hypothesized that PV, MF, and chronic myeloid leukemia (CML) were not separate entities but rather different manifestations of the same underlying problem: a myelostimulatory factor causing the bone marrow to overproduce cells. Critically, he noted that these diseases often overlapped or transformed into one another. For instance, a patient with polycythemia vera might eventually develop “spent-phase” myelofibrosis. He grouped them under the umbrella term myeloproliferative disorders (MPDs).
The undiscovered stimulus Dr. Dameshek hypothesized in 1951 was finally identified more than half a century later. The discovery of the JAK2 mutation in 2005 is considered the Big Bang of modern hematology. It transformed myeloproliferative neoplasms (MPNs) from poorly understood clinical syndromes into molecularly defined blood cancers. In a rare occurrence in the scientific community, four independent research groups (led by teams in France, the UK, Switzerland, and the USA) published the discovery of the same mutation within a few months of each other. They identified a single point mutation in the Janus Kinase 2 gene (JAK2), specifically a substitution of phenylalanine for valine at codon 617 (JAK2V617F), with each paper shedding additional light:
The French Team (James et al. Nature): Demonstrated that the mutation induces cytokine-independent growth in cells, mimicking the disease’s hyperproliferative nature.
The UK Team (Baxter et al. Lancet): Identified the mutation in 97% of PV patients and about half of those with ET and MF, confirming it as a common thread across Dr. Dameshek’s unified categories.
The Swiss Team (Kralovics et al. NEJM): Noted the transition from heterozygosity (one copy) to homozygosity (two copies) in some patients, explaining why some cases (like PV) appear more aggressive than others.
The US Team (Levine et al. Cancer Cell): Validated JAK2 as a drugable target, providing the first major evidence that inhibiting this specific protein could potentially treat the disease.
The breakthrough was enabled by a technological convergence. The human genome had been sequenced and high-throughput sequencing of the kinome (the set of all kinases) became possible. The story isn’t just that they found the same thing, but how the ‘V617F quartet’ each arrived at a related conclusion in different ways:
The French Team: They used a bottom-up biochemical approach. They noticed that cells from PV patients grew even when you blocked every other growth factor except for the JAK-STAT pathway. They essentially followed the smoke until they found the fire at JAK2 codon 617.
The Swiss Team: They used a top-down genetic approach called Uniparental Disomy (UPD) mapping. They looked at Chromosome 9, where they noticed many patients had two identical copies of a specific region from one parent (rather than one from each). They realized the “bad” mutation was likely hidden in that duplicated section and JAK2 was right in the middle of it.
The UK Team: This was a clinical-genomic powerhouse effort. They screened hundreds of patient samples from the Cancer Genome Project, looking specifically at kinases already known to be involved in blood production.
The US Team: They used a high-throughput sequencing screen of the tyrosine kinome. They systematically sequenced the most likely candidate genes across many patients until the JAK2V617F mutation appeared as a glaring, repeated hit.
Before this discovery, the field of MPNs was relatively niche. Ross Levine (one of the US lead authors) famously noted that at the 2004 ASH (American Society of Hematology) annual meeting, the MPN session was held on a Tuesday morning in a small room with about 50 people. After these four papers were published in early 2005, the field exploded. By the 2005 ASH meeting, the sessions were packed into massive ballrooms with thousands of attendees.
In science, being “scooped” can be devastating for a career or a lab’s funding. Once it became clear that multiple groups were breathing down the neck of the same discovery, the journals (Nature, The Lancet, NEJM, and Cancer Cell) moved with uncharacteristic speed. Rather than one group winning and the others losing, the simultaneous publication acted as a global confirmation that the finding was robust and undeniably true. Since these groups published within weeks of each other, they are almost always cited together in scientific literature as a block, ensuring that all four teams receive credit for ushering in the era of precision medicine for blood cancers.
Following the 2005 JAK2 breakthrough, researchers hypothesized that if JAK2 was mutated, perhaps the receptors that JAK2 binds to were also mutated. In 2006, research groups at Broad Institute/Harvard and the Mayo Clinic published papers in PLoS Medicine and Blood identifying mutations in the Thrombopoietin Receptor (MPL). They found that a specific spot in the protein, tryptophan at position 515, was frequently mutated to leucine (W515L) or lysine (W515K). MPL is the receptor for thrombopoietin, the hormone that stimulates platelet production. The mutation changes the receptor’s shape so that it behaves as if it is constantly activated, even when no thrombopoietin is present. This keeps the JAK2 protein attached to it in a state of permanent activation. MPL mutations are rare, found in only 5-10% of MF and ET cases, and virtually never in PV.
In 2008, the World Health Organization (WHO) formally changed the name from “myeloproliferative disorders” to “myeloproliferative neoplasms” (MPNs), officially recognizing MF and PV as blood cancers. This reclassification was much more than a semantic update; it was a fundamental shift in how the medical community viewed these diseases. Before 2005, the term “disorder” was used because doctors weren’t entirely sure if these were true cancers or simply over-enthusiastic bone marrow reactions. Conditions like PV and ET were often seen as chronic, manageable issues that someone lived with, unlike the aggressive nature of acute leukemias. The identification of JAK2 mutations suggested that these were clonal diseases. This means they originate from a single rogue stem cell that has acquired a genetic mutation giving it a survival advantage, the epitome of a neoplasm (a new, abnormal growth of tissue).
The 2008 WHO update also took the wild west of clinical diagnosis and turned it into a rigorous checklist. To be diagnosed with an MPN like PV, a patient now had to meet specific Major and Minor criteria, heavily weighted toward molecular testing. For the first time, testing for the JAK2V617F mutation (or similar markers) became a Major Criterion. If you had the mutation and a high red blood cell count, the diagnosis was almost certain. The WHO mandated a closer look at the architecture of the bone marrow. Pathologists began looking for specific clusters of megakaryocytes (the cells that make platelets) which are a hallmark of MPNs.
Recognizing these as neoplasms changed the urgency of the field in three key ways:
Pharma/Biotech Investment: Labeling a condition as a “neoplasm” or “cancer” often opens up different regulatory pathways and incentive structures for drug development. This helped pave the way for the clinical trials that led to the approval of JAK inhibitors.
Risk Stratification: Doctors began focusing more on leukemic transformation.”Since these were now recognized as cancers, the goal shifted from just managing symptoms to preventing the disease from evolving into Acute Myeloid Leukemia (AML).
Access to Care: The “cancer” designation often improved insurance coverage for expensive specialized treatments and made it easier for patients to access oncology centers and clinical trials that were previously reserved for more traditional malignancies.
Five years later in 2013, researchers uncovered a third driver mutation of MPNs. At the time, roughly 30% of MF and ET patients lacked JAK2 or MPL mutations, suggesting that another identified factor was a play. Two teams, led by Robert Kralovics (Vienna) and Tony Green (Cambridge), used Whole Exome Sequencing to scan the entire genetic code of JAK2-negative patients and identified mutations in Calreticulin (CALR). They published their results simultaneously in NEJM (Nangalia et al. NEJM, Klampfl et al. NEJM). CALR normally lives inside the cell (endoplasmic reticulum) helping proteins fold. However, the mutated CALR develops a pathological attraction to the MPL receptor. It binds to MPL inside the cell and escorts it to the surface, holding it in an active position. They identified CALR mutations in 70-84% of patients with non-mutated JAK2 and indicated that these mutations occur early in the disease’s clonal evolution. Beyond identifying the mutation, they highlighted that patients with CALR mutations had a lower risk of thrombosis and better overall survival compared to those with JAK2 mutations.
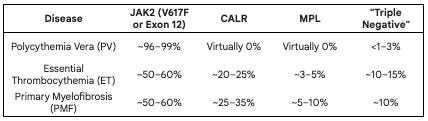
The prevalence of the three driver mutations (JAK2, MPL, CALR) varies significantly depending on which specific myeloproliferative neoplasm (MPN) is being discussed. Essentially, these mutations are mutually exclusive (a patient rarely has more than one), and together they account for approximately 90% of all classical MPN cases.
Polycythemia Vera is nearly 100% JAK2-driven: While about 95% of PV patients have the V617F mutation, the remaining few usually have mutations in JAK2 Exon 12. If a patient has high red blood cell counts but lacks any JAK2 mutation, clinicians typically look for secondary causes (like lung disease or smoking) rather than an MPN.
The CALR/MPL Split: You will notice that CALR and MPL mutations are essentially non-existent in PV. They are almost exclusively found in ET and MF, where the primary problem is platelet overproduction or marrow scarring rather than red cell excess.
Prognostic Value: These numbers aren’t just for diagnosis; they dictate the prognosis. For example, in Myelofibrosis, a patient with a CALR mutation (specifically Type 1) generally has a significantly longer median survival compared to a patient who is “Triple Negative” or carries a JAK2 mutation.
The Triple Negative Gap: About 10-15% of ET and PMF patients lack all three drivers. These cases are the current focus of intense research. In some of these “triple-negative” patients, deep sequencing reveals “atypical” or “non-canonical” mutations in JAK2 or MPL that standard tests missed.
Hitting the JAK-pot
The development of JAK inhibitors is a story of accelerated evolution in medicine. Within just six years of the genetic discovery of JAK2 in 2005, the first targeted therapy was approved by the FDA, a remarkably fast timeline for drug development. Even before the JAK2 mutation was identified in 2005, researchers knew that the JAK-STAT (Janus Kinase - Signal Transducer and Activator of Transcription) pathway was the master regulator of blood cell production. Scientists had already been experimenting with broad kinase inhibitors in other cancers. Once the JAK2V617F mutation was confirmed as the driver, pharmaceutical companies shifted from general research to a targeted race for a JAK2-specific molecule.
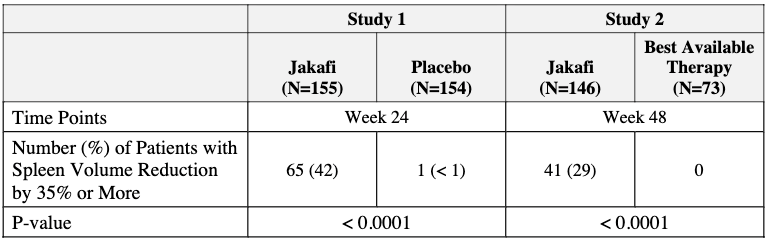
Incyte Corporation led the charge with a small molecule called INCB018424, later known as ruxolitinib and marketed as Jakafi by Incyte within the United States and Jakavi by Novartis outside the U.S. (Europe, Canada, etc.). Early in development, researchers realized ruxolitinib didn’t just help patients with the JAK2 mutation, it helped almost everyone with MF. This is because even “Triple Negative” patients have hyperactive JAK signaling. Two large Phase 3 trials (COMFORT-I and COMFORT-II) demonstrated that ruxolitinib dramatically reduced spleen size and improved debilitating symptoms (night sweats, fatigue, bone pain) compared to placebo or best available therapy (see below). In 2011, ruxolitinib (brand name Jakafi) became the first FDA-approved treatment for Myelofibrosis. It was later approved for Polycythemia Vera in 2014.
The success of Jakafi sparked a gold rush, but the journey was not without its challenges. Fedratinib was the biggest competitor to ruxolitinib. However, in 2013, the FDA placed a clinical hold on its development due to reports of Wernicke’s encephalopathy (a serious brain condition) in some patients. The drug languished for years before being rescued by Impact Biomedicines (later acquired by Celgene/Bristol-Myers Squibb) and was finally approved in 2019 and marketed as Inrebic after a re-analysis of safety data. Regulators found the incidence of WE was actually very low (approximately 1.3%) and manageable with thiamine monitoring. Inrebic offered the first second-line option for patients who were resistant or intolerant to Jakafi, specifically targeting the JAK2 and FLT3 kinases.
Recent years have seen the development of second-generation inhibitors designed to solve the limitations of Jakafi, specifically the dose-limiting toxicities of anemia and thrombocytopenia that left nearly 40% of myelofibrosis patients underserved:
Pacritinib (Vonjo, 2022): Developed specifically for patients with cytopenic myelofibrosis (those with very low platelet counts, <50,000/μL), who previously had no safe treatment options. Unlike its predecessors, Vonjo inhibits JAK2 and IRAK1 without significantly inhibiting JAK1. This JAK1-sparing profile allows it to shrink the spleen without causing the same degree of bone marrow suppression.
Momelotinib (Ojjaara, 2023): This drug was unique because it inhibits not just JAK2, but also ACVR1. This dual action helps reduce levels of hepcidin, effectively treating the anemia that often plagues MF patients. By inhibiting ACVR1, Ojjaara lowers hepcidin levels, unlocking the body’s iron stores for red blood cell production. The pivotal Phase 3 MOMENTUM trial showed that patients on Ojjaara were significantly more likely to become transfusion independent compared to those on other therapies.
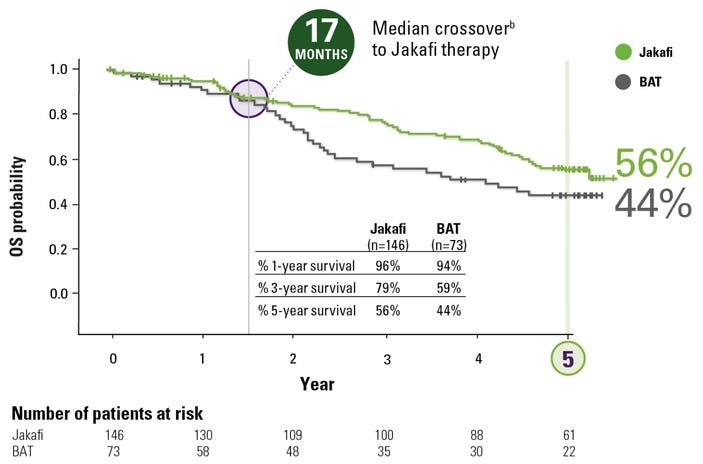
In the pre-JAK inhibitor era (before 2011), the median survival for high-risk myelofibrosis was often cited at roughly 2 to 3 years. Today, for patients treated with modern therapies, that trajectory has significantly shifted. The most robust numbers come from the long-term follow-ups of the COMFORT-I and COMFORT-II trials for Jakafi. Pooled analysis of these trials showed a reduction in the risk of death by approximately 30% to 35% compared to placebo or “Best Available Therapy” (BAT). In COMFORT-II, the survival probability at 3 years was 79% for those on Jakafi, compared to only 59% for those on traditional therapies. Since many patients in the control groups eventually switched to the JAK inhibitor (crossover), the “true” survival benefit was initially hidden. When researchers adjusted for this crossover, the estimated 3-year survival for the control group dropped to 31% versus 78% in the Jakafi arm, highlighting a wide gap in favor of the JAK inhibitor. The numbers also revealed a fascinating biological link: patients who achieved a ≥35% reduction in spleen volume had significantly longer survival than those who did not. This suggested that shrinking the spleen wasn’t just about comfort, it was a surrogate for changing the disease’s course.
The survival improvement in PV is even more dramatic, largely because the disease is more indolent and management of blood thickness (hematocrit) has become highly standardized. The foundational 1962 study describing the natural history of PV reported that patients who received treated with phlebotomy alone had a median survival of about 3.5 years. In a more recent survival model presented at ASH 2024, median survival is approximately 24-28 years in patients <60 years old.
The numbers support the claim that survival in MF and PV are improving due to two main factors:
Reduction in Thromboembolic Events: By better controlling hematocrit (in PV) and reducing inflammatory cytokines (in MF), the immediate “killer” (blood clots and strokes) has been significantly curbed.
Phenotype-Specific Dosing: Second generation JAK inhibitors (Vonjo, Ojjaara) allow patients with low blood counts to stay on therapy longer. Previously, these patients would have had to stop treatment, leading to rapid disease progression.
Jacking up JAK2
The first and second generation of targeted therapies for Myelofibrosis comprises Type I JAK2 inhibitors. These agents are designed to target the protein in its “active” state, technically referred to as the DFG-in conformation. Despite their success in improving quality of life, Type I inhibitors leave some room for improvement:
Lack of Molecular Remission: These agents excel at reducing spleen volume and inflammatory symptoms but can leave behind treatment-resistant clones that lead to relapsed disease.
High Discontinuation Rates: Clinical data indicates that most patients discontinue Type I therapy within 2 to 3 years due to a loss of response, disease progression, or treatment-limiting adverse events.
Persistent Bone Marrow Fibrosis: After 24 months on Jakafi, only 15% of patients showed a histopathologic improvement in fibrosis grade, while the vast majority simply stabilized or continued to worsen.
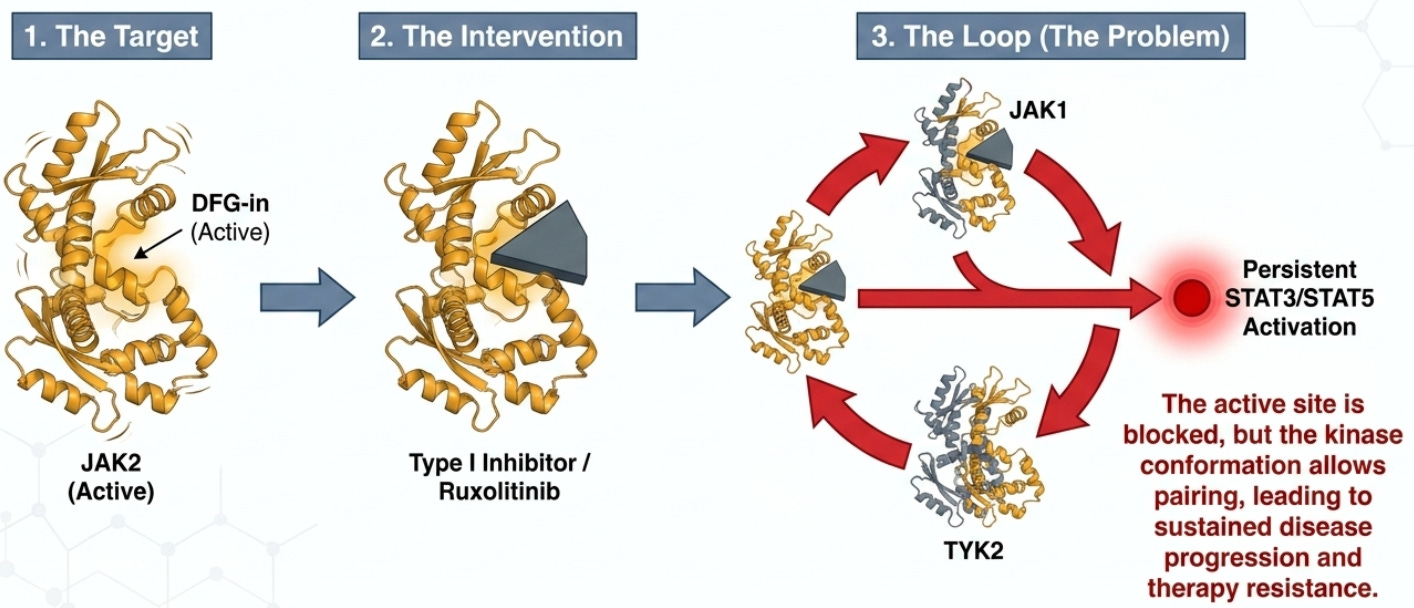
The primary reason for these limitations lies in the biochemical “loophole” inherent in Type I binding, which allows cancer cells to hide in plain sight through alternative signaling architectures. When a Type I inhibitor binds to JAK2 in its active DFG-in state, it paradoxically stabilizes the protein in a way that facilitates heterodimerization. In this state, inhibited JAK2 can form complexes with other members of the JAK family, specifically JAK1 or TYK2. In other words, Type I inhibitors jam the lock while the door is open, but the hinges (other JAKs) still let the signal through. Type II inhibitors are designed to lock the door before it ever opens.
This molecular bypass allows for the continued, or persistent, activation of downstream effectors STAT3 and STAT5. Consequently, the malignant cells remain viable and continue to drive disease progression despite the presence of the inhibitor. This phenomenon, known as clonal persistence, necessitates a shift toward targeting the protein in its resting state. To overcome this resistance, the next generation of drug design has shifted focus toward targeting the protein’s inactive architecture.
The shift to Type II JAK2 inhibitors represents a next step in MPN therapy. These agents target the protein in its inactive or resting conformation, known as DFG-out. Ajax Therapeutics’ lead program AJ1-11095 is a first-in-class Type II inhibitor developed through advanced computational structure-based design. Its primary mechanism of action is to bind and lock JAK2 in the DFG-out state, which prevents the formation of the JAK2/JAK1 and JAK2/TYK2 complexes that drive clonal persistence. By maintaining the protein in an inactive state, Type II inhibitors aim to achieve what Type I agents cannot: the systematic reduction of the disease-driving clone. The transition from theoretical design to clinical application is supported by robust laboratory evidence demonstrating a fundamental change in the disease environment.
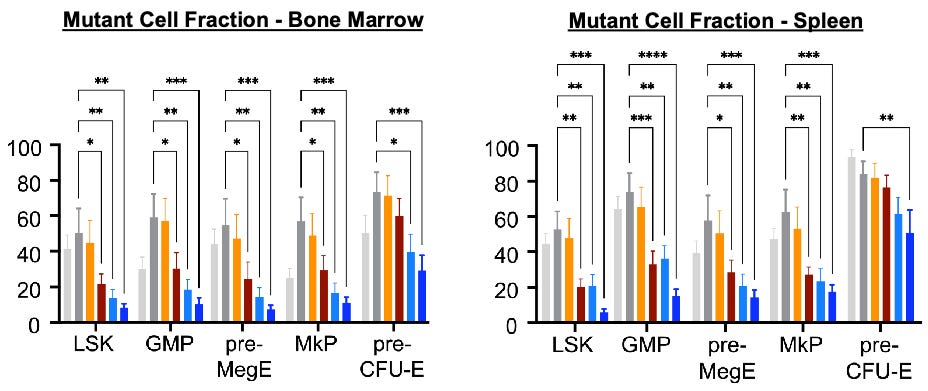
Preclinical evaluations of AJ1-11095 and its predecessor, AJ1-10502, utilized sophisticated mouse models, including the hMPLW515L adoptive transfer model and the dual Dre/Cre-recombinase Jak2VF model. These studies provide the foundation for the “3 Pillars of Efficacy” required for disease modification:
Reduction of Mutant Allele Burden: Compared to Type I agents, AJ1-11095 demonstrated a significant reduction in the percentage of mutant cells within the bone marrow and spleen (see below), specifically targeting hematopoietic stem cells (HSCs) and granulocyte-macrophage progenitors (GMPs).
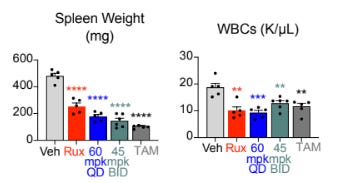
Spleen and Symptom Control: Preclinical models showed dramatic and dose-dependent reductions in splenomegaly and leukocytosis, often approaching levels seen with genetic deletion of the JAK2 mutant.
Reduction of Fibrosis: Most critically, histopathologic analysis through reticulin staining revealed lower bone marrow fibrosis, suggesting suggesting the potential for restoration of the healthy marrow microenvironment.
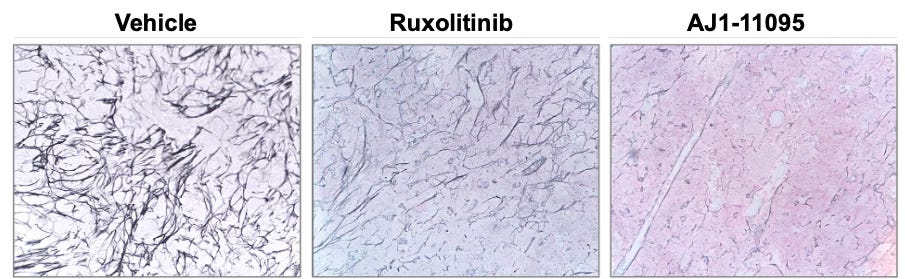
While preclinical mouse models hint at these ‘3 Pillars of Efficacy,’ it remains to be seen if these results will translate into human subjects in the ongoing AJX-101 trial. A distinguishing feature of AJ1-11095 is its selectivity for Type II, which is paramount for minimizing off-target toxicities. Biochemical assays confirm that AJ1-11095 is:
780x more selective for JAK2 over JAK1
>3840x more selective for JAK2 over JAK3
150x more selective for JAK2 over TYK2
The clinical utility of AJ1-11095 is currently being evaluated in AJX-101, a Phase 1, multicenter, open-label dose-escalation and expansion study. This trial targets a high-unmet-need population: adults with primary or post-ET/PV Myelofibrosis who have failed or relapsed after prior Type I therapy and possess DIPSS Intermediate-2 or High-risk disease. The study employs a conventional 3+3 design for initial cohorts, with subsequent dose escalation guided by a modified Fibonacci sequence. The starting dose was established at 25 mg once daily. The trial seeks to define the Maximum Tolerated Dose (MTD) and the Recommended Phase 2 Dose (RP2D). Key secondary endpoints include a 35% reduction in spleen volume (SVR35) and a 50% reduction in Total Symptom Score (TSS50) at 24 weeks. AJ1-11095 recently received Orphan Drug Designation from the FDA in December 2025 and Eli Lilly expects initial proof-of-concept data later in 2026.
The ability to target the “inactive” DFG-out state (Type II) represents a potential improvement in the treatment of myeloproliferative neoplasms (MPNs) like myelofibrosis (MF) and polycythemia vera (PV). While the first decade of JAK2 inhibition focused on symptomatic management, the emergence of Type II inhibitors like AJ1-11095 offers a potential path toward disease modification. An inhibitor that can disrupt the biochemical mechanisms of clonal persistence, could ultimately result in deeper molecular remissions and improved long-term outcomes for patients.
The Rise of Ajax Therapeutics
The story of Ajax Therapeutics is a textbook example of how academic breakthroughs, specialized venture capital, and cutting-edge computational chemistry can converge to disrupt an established drug class. The company’s trajectory has shifted from a stealth startup to a major acquisition target, culminating in a $2.3 billion deal with Eli Lilly in April 2026.
Ajax Therapeutics was established based on pioneering research from the laboratory of Dr. Ross Levine at Memorial Sloan Kettering Cancer Center (MSKKC). Dr. Levine, a world-renowned expert in myeloproliferative neoplasms (MPNs), co-founded the company alongside other experts in oncology and drug discovery to translate insights into the molecular drivers of blood cancers into targeted therapies.
In 2014, Dr. Levine published a landmark paper in the journal Blood showing that Type II inhibitors (which bind to the inactive state of the JAK2 enzyme) could theoretically offer deeper, more durable responses. This scientific insight sat in the academic realm until the right technical partnership could be formed to solve the massive chemistry challenge of making these molecules selective. In this paper, Levine’s team used an innovative “knock-in/knock-out” mouse model to prove that completely removing the JAK2 V617F mutation leads to a more pronounced suppression of the disease compared to Type I inhibitors (we discussed this in the earlier section). While many researchers were looking for new targets, this paper proved that JAK2 was still the right target, it just wasn’t being hit hard enough. This realization led directly to the pursuit of Type II inhibitors (ie. those developed by Ajax), which bind to the inactive state of the protein and prevent persistent JAK-STAT signaling from occuring.
Ajax Therapeutics was formally founded in 2019 by Dr. Levine, Olli Silvennoinen, and Martin Vogelbaum (who transitioned from an investor to the CEO role). Unlike many startups that spend years in the wilderness, Ajax had a running start due to a unique founding partnership:
Schrödinger, Inc.: Instead of building a traditional wet lab immediately, Ajax relied heavily on Schrödinger’s computational drug discovery platform. This allowed them to “digitally” design and test thousands of molecules to find a Type II inhibitor that was highly selective for the JAK2 V617F mutation—something previously considered nearly impossible.
Strategic Backing: From the beginning, Eli Lilly was a founding strategic investor. This is a rare setup where a Big Pharma company helps build the startup from the ground up, providing a clear eventual exit path.
By 2021, Ajax closed a $40 million Series B led by HealthCap. This phase was about building the team, hiring experts like Dr. Craig Masse (formerly of Nimbus) and Dr. Christina Riordan, to move from digital designs to actual manufacturing and regulatory filings.
In April 2024, the company reached escape velocity with a $95 million Series C led by Goldman Sachs Alternatives. This capital was used to launch the AJX-101 Phase 1 trial for their lead candidate, AJ1-11095. This trial targeted the most difficult patient population: those who had already failed or stopped responding to first-generation JAK inhibitors.
The story reached its climax on April 27, 2026. Eli Lilly, seeing the early Phase 1 data and wanting to solidify its oncology portfolio, exercised its position as a strategic partner to fully acquire the company.
Conclusion
From Dr. Gustav Heuck’s first descriptions in 1879 to the “Big Bang” of the 2005 JAK2 discovery, the story of MPNs has been one of slowly unlocking a molecular puzzle. Ajax Therapeutics represents the latest, and perhaps most precise, piece of that puzzle. By solving the chemistry challenge that long plagued Type I inhibitors, Ajax has bridged the gap between Dr. Ross Levine’s 2014 hypothesis and a tangible therapy. As Eli Lilly takes the reins, the narrative of myelofibrosis moves from a history of accidental observations to a future of intentional, structure-based design. The “running start” that began in a MSKCC lab is now a sprint toward a potentially new standard of care for patients with myelofibrosis.
To contact us, please send us an email at biotechreadout@gmail.com
Disclaimers
Investigational Status Disclaimer
The therapeutic candidates discussed in this newsletter are currently in clinical development and have not been approved for commercial sale by the U.S. Food and Drug Administration (FDA), the European Medicines Agency (EMA), or other global regulatory authorities. Their safety and efficacy have not been established. References to pipeline products and ongoing clinical trials involve significant risks and uncertainties. Statements regarding the potential safety, potency, or efficacy of investigational drugs reflect current hypotheses and are not a guarantee of future performance or regulatory clearance. The outcome of clinical trials is inherently unpredictable, and clinical results from earlier stages may not be predictive of results in later, larger-scale trials. This article includes discussion of AJ1-11095, an investigational compound not yet approved by the FDA. Statements regarding its mechanism are based on early-stage data and are not predictive of final clinical outcomes.
No Medical Advice Disclaimer
This newsletter is for informational and educational purposes only. The content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or another qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read in this publication.
No Patient-Provider Relationship Disclaimer
The information provided in this newsletter is for educational and analytical purposes only. Receipt of this information, or any interaction with this content, does not create a physician-patient, pharmacist-patient, or any other professional-provider relationship between you and the authors or publishers. This newsletter should not be used as a substitute for a personal consultation with a qualified healthcare professional.
Forward-Looking Statements Disclaimer
This newsletter contains “forward-looking statements” regarding future events, including clinical trial timing, regulatory milestones, and projected market performance. These statements are based on current expectations and assumptions that are subject to significant risks and uncertainties. Actual results may differ materially from those expressed or implied. We undertake no obligation to update these statements as a result of new information or future developments.
Third-Party Links & Content Disclaimer
This newsletter contains links to third-party websites, including clinical trial registries and corporate presentations. Biotech Readout does not endorse, guarantee, or assume responsibility for the accuracy or reliability of any information offered by third-party providers.
Errors and Omissions Disclaimer
While we strive for technical accuracy, the information in this newsletter is provided on an “as is” basis with no guarantees of completeness, accuracy, or timeliness. Biotech Readout assumes no liability for any errors or omissions in the content of this publication.
Non-Endorsement Disclaimer
Any reference to specific commercial products, processes, or services by trade name, trademark, or manufacturer does not constitute or imply an endorsement or recommendation by the author. All trademarks are the property of their respective owners.
No Investment Advice Disclaimer
This newsletter is for informational purposes only and does not constitute financial, investment, or legal advice. The author is not a registered investment advisor. You should consult with a professional financial advisor before making any investment decisions. The biotechnology sector is highly volatile; past performance is not indicative of future results.
Conflict of Interest Disclaimer
The author of this newsletter maintains a position of independence. At the time of publication, the author holds no direct financial interest, equity, or options in any of the companies mentioned in this report. No compensation has been received from any third party to feature or analyze specific therapeutic candidates or corporate entities.