
First approval in recently diagnosed T1D, gene therapy rewrites HAE, and more
Weekly Readout #10: Week ending June 19, 2026

Disclaimer: This newsletter is for educational and informational purposes only and does not constitute medical, investment, or financial advice, nor does it establish a provider-patient relationship. Content may include forward-looking statements and discussions of investigational therapeutic candidates that are not FDA/EMA approved; their safety and efficacy remain unestablished and clinical outcomes are unpredictable. While we strive for accuracy, all information is provided as is without guarantees. This newsletter is independent, and the author holds no financial positions in the companies mentioned nor receives third-party compensation for this coverage. Please find a complete version of our disclaimers at the bottom of this article and on our About page.

Press ⬇️ to go down to a section and ⬆️ to go back up to the Table of Contents
Table of Contents
This week, we discuss:
China Biotech
U.S. Treasury Department Weighs Screening Investments in China Biotech ⬇️
Acquisitions ⬇️
Biogen to Acquire RayThera
Eli Lilly to Acquire 4E Therapeutics
Approvals
Clinical Trial Data
China Biotech
U.S. Treasury Department Weighs Screening Investments in China Biotech
An Endpoints article published last night outlines a strategic escalation in Washington’s scrutiny of cross-border capital flows, specifically looking at how the U.S. Department of the Treasury is evaluating outbound investment screening protocols for the Chinese biotech sector. Expanding beyond original executive restrictions focused heavily on semiconductors, quantum computing, and specific AI infrastructure, the federal framework is actively assessing biotechnology as a critical dual-use and national security vertical. Two unnamed sources at Treasury stated, “biotech is likely to be included in coming draft regulations that will say which sectors are going to be subject to outbound investment screening.” The Treasury is weighing rules that would mandate disclosures or outright restrict U.S. venture capital, private equity, and joint-venture capital from funding Chinese entities engaged in advanced biomanufacturing, genomics, and synthetic biology.
In a piece that I wrote about 3 weeks ago entitled China Biotech: Feast or Famine?, I discussed the risks & rewards associated with deeper entanglement of U.S. and China Biotech. Chinese biotech has evolved from an era of fast-follower generics into a powerhouse of validated, highly competitive clinical assets, particularly in oncology and immunology. The Treasury’s push to screen investments represents a surgical attempt to prevent U.S. capital from building up foundational infrastructure within China that could, ultimately, hollow out U.S. infrastructure.

Acquisitions

Approvals
Sanofi - Tzield (anti-CD3 mAb) / T1D
In a historic development for diabetes care, the FDA granted accelerated approval to Sanofi’s Tzield (teplizumab-mzwv), making it the first-ever disease-modifying therapy approved for children and adolescents aged 8 to 17 years who have been recently diagnosed with Stage 3 Type 1 Diabetes (T1D). For over a century, treating Stage 3 T1D (the symptomatic phase where patients require daily insulin injections) has been entirely reactive, focusing on managing blood sugar after the pancreas has already suffered severe damage. Previously approved only to delay the onset of Stage 3 disease in patients with asymptomatic Stage 2 T1D, this new approval allows clinicians to intercept the disease after a clinical diagnosis. It aims to protect the remaining insulin-producing beta cells in the pancreas before they are completely destroyed by the body’s own immune system. The FDA’s decision was heavily anchored by data from the Phase 3 PROTECT study, a randomized, double-blind, placebo-controlled clinical trial involving 328 pediatric patients diagnosed with Stage 3 T1D within the preceding six weeks. The trial successfully met its primary goal, demonstrating that Tzield significantly improved and preserved beta cell function by substantially slowing down the decrease of average C-peptide levels compared to the placebo group.
Merck - Welireg (HIF-2α inhibitor) & Keytruda (anti-PD1 mAb) / ccRCC
The FDA granted a significant label expansion to Merck’s oncology portfolio, officially approving the combination of Welireg (belzutifan) and Keytruda (pembrolizumab). This milestone introduces the first-ever approved regimen combining a PD-1 inhibitor with a HIF-2α inhibitor for the adjuvant treatment of clear cell renal cell carcinoma (ccRCC). The FDA’s decision was fast-tracked via a priority review based on compelling data from the Phase 3 LITESPARK-022 trial, a global study involving 1,841 patients. At a pre-specified interim analysis, the Welireg combination demonstrated a 28% reduction in the risk of disease recurrence, metastasis, or death compared to Keytruda alone (HR = 0.72, P= 0.0003).
Clinical Trial Data
Intellia - Lonvo-z (KLKB1 KO) / Phase 3 (HAE)
The Phase 3 data for Intellia’s lonvoguran ziclumeran (lonvo-z) was published in NEJM and marks a watershed moment in genetic medicine, delivering the first-ever complete Phase 3 dataset for an in vivo CRISPR gene-editing therapy.
Hereditary Angioedema (HAE) is a rare, autosomal dominant genetic disorder where a deficiency or dysfunction in the C1 esterase inhibitor (C1-INH) protein leads to the unchecked activation of the contact pathway, driving hyper-production of the vasoactive peptide bradykinin, which triggers life-threatening vascular leakage and severe swelling. The standard of care is bifurcated into acute and preventative management: acute attacks are intercepted with on-demand therapies like intravenous C1-INH concentrates (Berinert), plasma kallikrein inhibitors (Kalbitor), or B2 receptor antagonists (Firazyr), while chronic long-term prophylaxis (LTP) relies on routine sub-Q infusions of C1-INH (Haegarda), oral small-molecule kallikrein inhibitors (Orladeyo), or monoclonal antibodies targeting activated kallikrein (Takhzyro). Delivered as a single intravenous lipid nanoparticle infusion, lonvo-z utilizes in vivo CRISPR/Cas9 gene editing to permanently knock out the liver’s KLKB1 gene, potentially potentially fixing the root cause of HAE. By selectively shutting down the production of prekallikrein (the upstream precursor to active kallikrein) this one-time intervention halts the continuous generation of downstream bradykinin, offering a functional cure by structurally preventing the vascular permeability that drives HAE attacks.
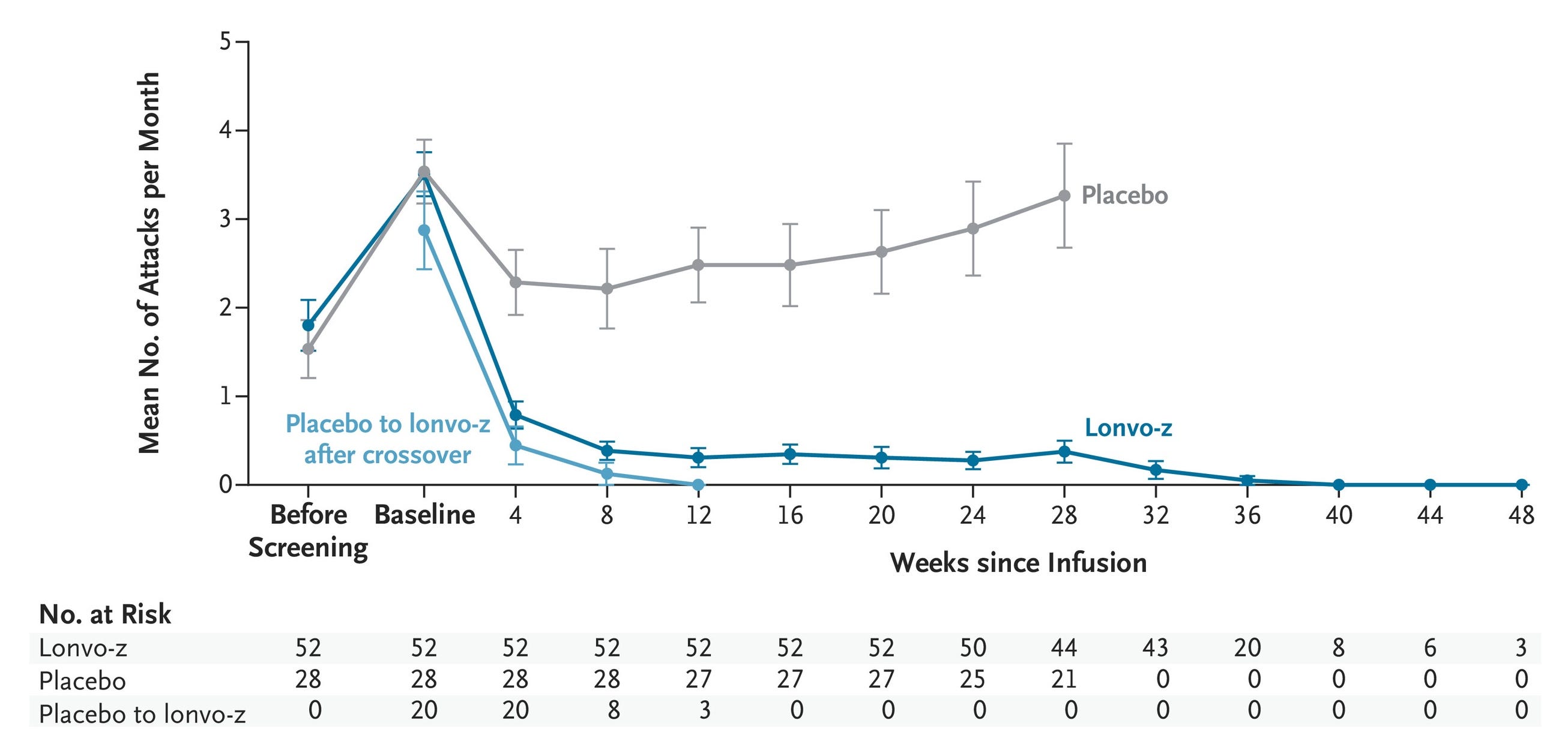
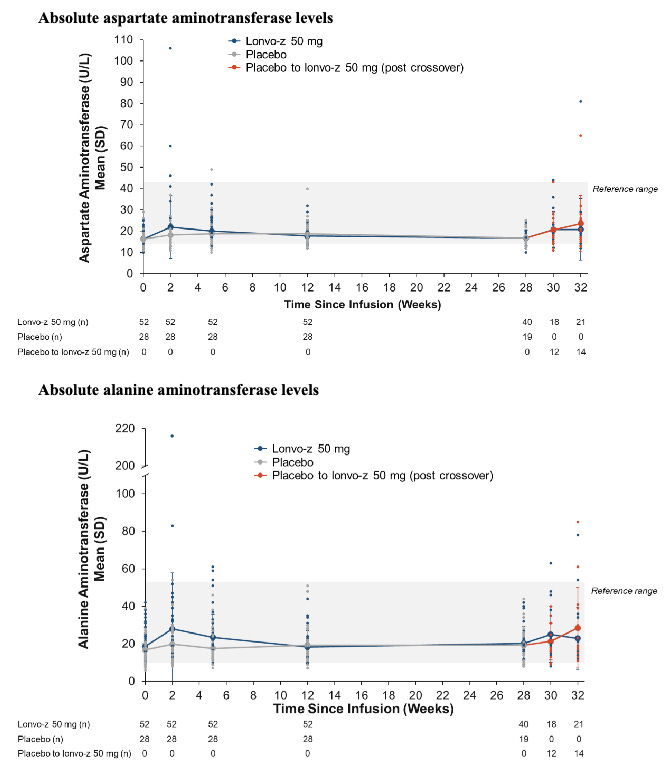
The global, randomized, double-blind, placebo-controlled Phase 3 HAELO trial (N=80) evaluating lonvo-z in Type I/II HAE patients established the first-ever Phase 3 validation of a systemic, in vivo CRISPR-Cas9 platform. The trial met its primary endpoint with a highly significant 87% reduction in mean monthly attacks (0.26 vs. 2.10 for placebo; P<0.0001), alongside a 91% reduction in moderate-to-severe episodes, enabling 62% of patients to achieve a completely attack-free status without long-term prophylaxis. On the safety front, the therapy was well-tolerated with no serious adverse events. The knock is that approximately 15% of patients experienced AST/ALT liver enzyme elevations. However, this transaminitis was entirely transient, asymptomatic, and self-limiting, reflecting a standard, class-wide characteristic of LNP clearance by hepatocytes. Ultimately, by delivering a uniform attack-rate reduction across 100% of treated patients, lonvo-z could minimize breakthrough anxiety and might transition HAE management away from a lifetime of chronic injections and toward a one-time, durable functional cure (counteracting the root cause of HAE).
Intellia initiated a rolling Biologics License Application (BLA) submission with the U.S. FDA in April 2026. The company plans to finalize the complete BLA submission package by H2 2026. Leveraging expedited regulatory designations, Intellia is targeting an FDA approval decision and potential commercial U.S. launch in H1 2027. Patients from the HAELO trial will continue to be monitored in long-term extension protocols to confirm the life-long durability of the KLKB1 gene knockout.
Sources: Intellia press release, NEJM paper
J&J - Talvey (GPRC5D x CD3 TCE) / Phase 3 (RRMM)
The Phase 3 data for Johnson & Johnson’s Talvey (talquetamab-tgvs) represents a major advance in hematologic oncology. Presented as a plenary abstract at the 2026 European Hematology Association (EHA) Congress, the results from the MonumenTAL-3 trial provide the first randomized, Phase 3 validation for moving a GPRC5D-targeted bispecific antibody combination into earlier-line treatment settings for multiple myeloma. The data was also published in NEJM.
Relapsed or refractory multiple myeloma (RRMM) occurs when malignant plasma cells evolve and escape frontline therapies (typically lenalidomide-based regimens), triggering bone marrow infiltration and clinical manifestations like anemia, hypercalcemia, and lytic bone lesions. While the current second-line standard of care relies on class-switching triplet regimens, such as pairing an anti-CD38 monoclonal antibody with a next-generation immunomodulatory drug (iMiD) or a proteasome inhibitor, Johnson & Johnson’s Talvey (talquetamab-tgvs) introduces a novel T-cell redirecting paradigm. As a first-in-class bispecific T-cell engager (TCE) targeting GPRC5D, Talvey simultaneously binds to CD3 on cytotoxic T cells and GPRC5D, an orphan receptor highly overexpressed on myeloma cells but absent on healthy B cells, thereby preserving baseline humoral immunity. When combined with Darzalex Faspro (daratumumab), the regimen drives anti-tumor synergy. Darzalex systematically de-bulks immunosuppressive cells and primes the marrow microenvironment, significantly amplifying Talvey’s targeted T-cell-mediated destruction of refractory clones.
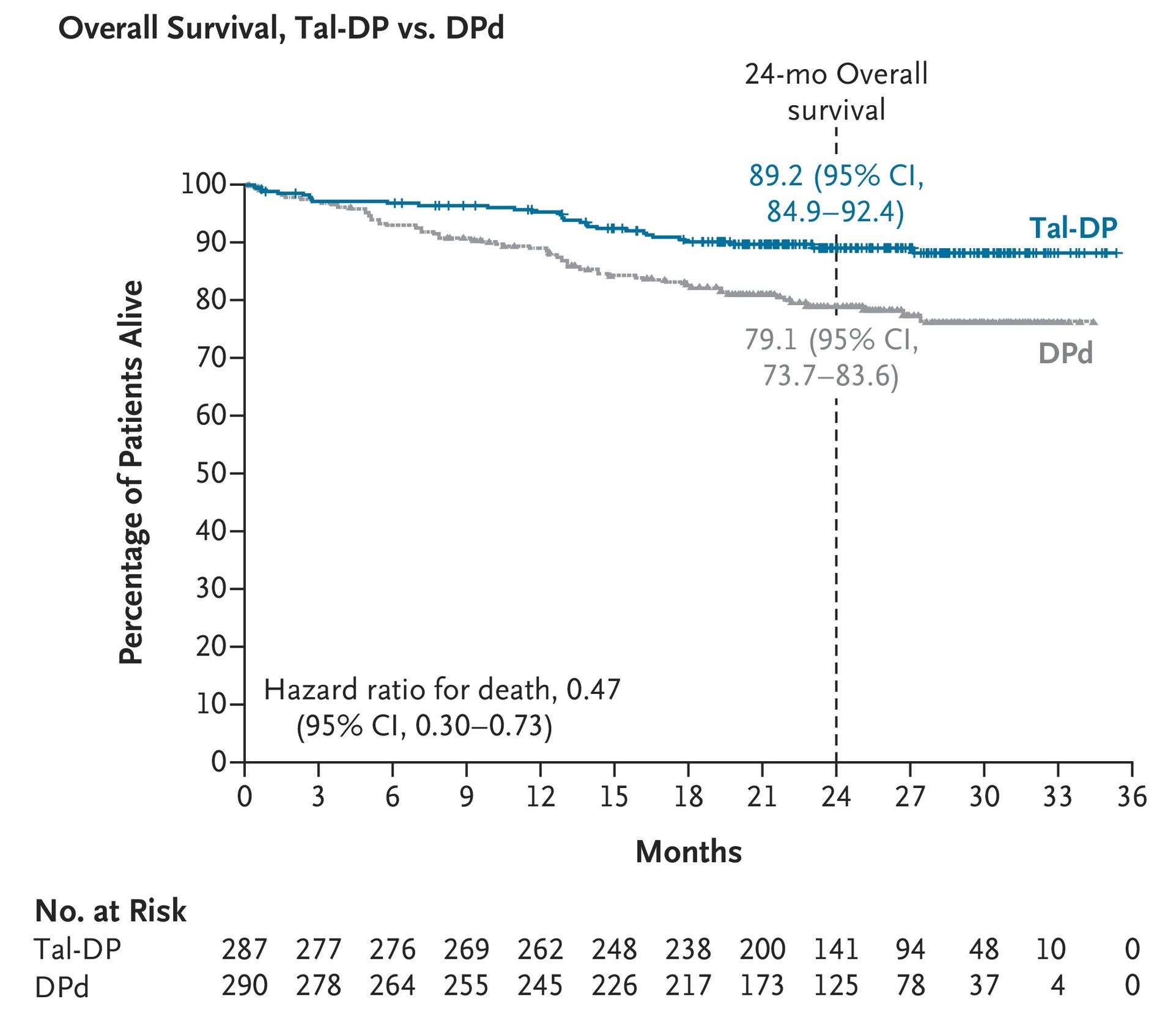
The global, randomized Phase 3 MonumenTAL-3 trial (N=864) evaluating Talvey (talquetamab-tgvs) combinations against standard-of-care DPd in early relapsed/refractory multiple myeloma (2L+) demonstrated unprecedented efficacy, breaking Talvey out of its late-line niche and potetnially establishing it as a premier frontline option, particularly for tough-to-treat lenalidomide-refractory patients. At a median follow-up of 24.6 months, both experimental arms demonstrated overwhelming, statistically significant superiority over the standard of care, showing a 67-72% reduced risk of disease progression or death (Tal-DP: HR = 0.28, P < 0.0001; Tal-D HR = 0.33, P < 0.0001) and a 49-53% reduced risk of death (Tal-DP: HR = 0.47, P = 0.0006; Tal-D HR = 0.51, P = 0.0015). The overall response rates (ORR) hovered around 88% for both Talvey arms (vs. 77.6% for DPd). Furthermore, the Talvey regimens almost doubled complete response rates (71.1% for Tal-DP and 69.0% for Tal-D versus 34.5% for standard of care), translating to an impressive MRD-negativity rate (52.3% Tal-DP and 46.3% Tal-D versus 15.9% for standard of care). While expected, low-grade GPRC5D-related skin and taste toxicities occurred frequently, the doublet configuration (Tal-D) presented an invaluable, highly potent iMiD-free treatment alternative that dramatically slashed the rate of severe Grade 3/4 infections (29.2% vs. 42.2% for standard DPd) to optimize patient quality of life.
Backed by this definitive dataset, Johnson & Johnson has already submitted a supplemental Biologics License Application (sBLA) to the U.S. FDA seeking approval for Talvey + Darzalex Faspro (with or without pomalidomide) in patients with RRMM who have received at least one prior line of therapy. J&J is preparing to aggressively position this combination to capture early-line market share, directly challenging established triplet and quadruplet sequencing strategies. Future clinical analysis will focus on how this GPRC5D-targeted combination can best be sequenced with or after BCMA-targeted CAR-T therapies (like Carvykti) and other T-cell engagers.
Sources: J&J press release, NEJM paper
Eli Lilly - AJ1-11095 (Type II JAK2 inhibitor) / Phase 1 (myelofibrosis)
The Phase 1 data presentation for Eli Lilly’s newly acquired AJ1-11095 (from Ajax Therapeutics) represents a major technological leap forward in treating myelofibrosis. Presented orally at the 2026 European Hematology Association (EHA) Congress by Dr. John Mascarenhas (Abstract S218), the initial readout establishes a compelling validation for the industry’s first clinical-stage Type II JAK2 inhibitor.

Myelofibrosis is a rare myeloproliferative neoplasm driven by somatic mutations (JAK2, CALR, or MPL) that hyperactivate the JAK/STAT signaling pathway, triggering a pro-inflammatory cytokine storm that results in progressive bone marrow fibrosis, severe cytopenias, and extramedullary hematopoiesis presenting as profound splenomegaly. While the current intermediate-to-high risk standard of care relies heavily on approved Type I JAK inhibitors (ruxolitinib, fedratinib, pacritinib, or momelotinib) these conventional agents only bind to the active conformation of the JAK2 kinase, failing to eradicate the underlying mutant clone and frequently inducing resistance or “clonal persistence” via adaptive pathway heterodimerization. Eli Lilly’s AJ1-11095 introduces a novel structural paradigm as a first-in-class oral Type II JAK2 inhibitor designed via computational chemistry to bind selectively to the inactive, DFG-out conformation of the kinase. By structurally stabilizing this inactive state, AJ1-11095 is designed to prevent both JAK2 homodimerization and cross-talk heterodimerization with JAK1 or TYK2, potentially unlocking deeper, more durable signaling suppression capable of fundamentally overcoming the molecular workarounds that drive Type I treatment failure.
The global, open-label Phase 1 AJX-101 trial (N=23) utilizing a traditional 3+3 dose-escalation design across five once-daily oral cohorts (25–125 mg) investigated AJ1-11095 as a potentially new second-line salvage paradigm for intermediate-2 or high-risk primary/secondary myelofibrosis patients who had failed or proved completely refractory to at least one prior Type I JAK inhibitor. AJ1-11095 achieved a 70% SVR35 rate (16/23 patients) and a parallel 70% TSS50 rate by Week 12, outperforming historical 2L benchmarks (0–32%). Crucially, the candidate demonstrated the first clear evidence of true molecular remission and disease modification in this setting, with 91% of patients achieving a reduction in mutant Variant Allele Frequency (VAF) across JAK2, MPL, or CALR clones, including a ≥50% VAF reduction in 35% of patients by Week 24. On the safety front, the small molecule had zero dose-limiting toxicities or treatment discontinuations. Expected hematologic class effects like anemia (52% Grade ≥3) and thrombocytopenia (30% Grade ≥3) were effectively managed via dose adjustments, while a 35% incidence of ALT/AST liver enzyme elevations proved entirely transient, asymptomatic, and self-limiting.
Following the completion of the escalation phase, Eli Lilly has selected the 75 mg once-daily dose as the candidate for initial dose expansion. The program has rapidly transitioned into recruiting a larger 2L myelofibrosis expansion phase to solidify these registrational metrics. Backed by their recent $2.3 billion acquisition of Ajax Therapeutics in April 2026, Lilly plans to expand the clinical footprint of AJ1-11095 into treatment-naïve (1L) myelofibrosis as well as high-risk polycythemia vera.
Sources: Eli Lilly press release, Eli Lilly to Acquire Ajax
Edgewise - EDG-7500 (allosteric cardiac myosin inhibitor) / Phase 2 (oHCM & nHCM)
The top-line data readout for Edgewise Therapeutics’ EDG-7500 represents a new development in the hypertrophic cardiomyopathy (HCM) landscape, delivering strong Phase 2 data that sets up a direct late-stage challenge to established cardiac myosin inhibitors (CMIs). Importantly, this it the first major clinical update from Edgewise after the sale of their muscular dystrophy business to Servier for up to $2.65 billion ($1.55 billion upfront + up to $1.1 billion in regulatory and commercial milestone payments) and focus on cardiovascular.
Servier to Acquire Edgewise's Muscular Dystrophy Medicine

A contrarian bet on skeletal myosin pays off
Hypertrophic Cardiomyopathy (HCM) is a genetic disorder marked by sarcomeric mutations that cause myocardial hypercontractility and fibrosis. Obstructive HCM (oHCM) occurs when septal hypertrophy drives systolic anterior motion (SAM) of the mitral valve to create a dynamic left ventricular outflow tract (LVOT) obstruction. Nonobstructive HCM (nHCM) is primarily governed by severe diastolic dysfunction and elevated filling pressures. While oHCM standard-of-care management relies on negative inotropes and first-generation cardiac myosin inhibitors (CMIs) like mavacamten or aficamten to resolve the physical gradient, nHCM treatment remains strictly limited to palliative diuretics, underscoring a vast unmet need for therapies that improve relaxation without sacrificing systolic output. Edgewise’s EDG-7500 addresses this gap as an oral, selective cardiac sarcomere modulator that is designed to uniquely act during early diastole to slow contraction velocity and enhance myocardial relaxation. By precisely targeting the initial millisecond phase of early systole, EDG-7500 aims to alleviate LVOT obstruction in oHCM and ease stiffness in nHCM without triggering the class-wide drop in overall contractile force, successfully preserving baseline Left Ventricular Ejection Fraction (LVEF) and avoiding the severe ventricular safety risks of older CMIs.
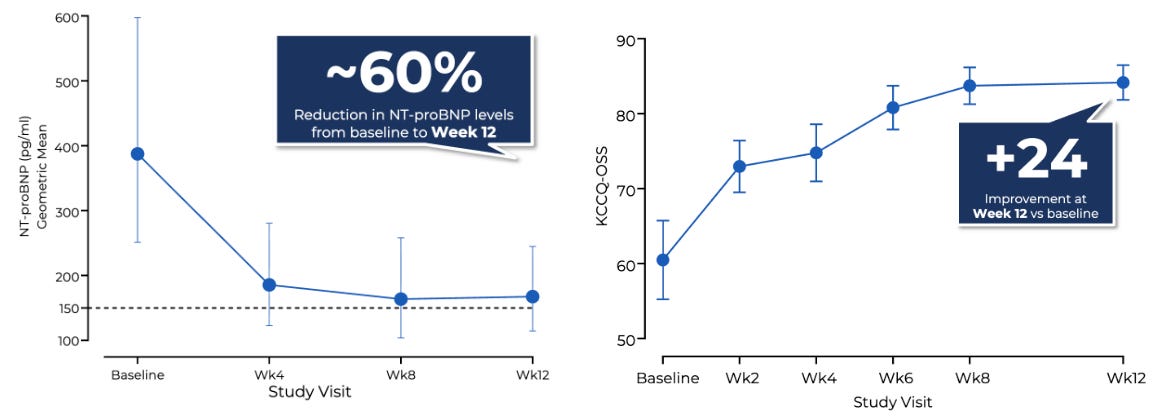
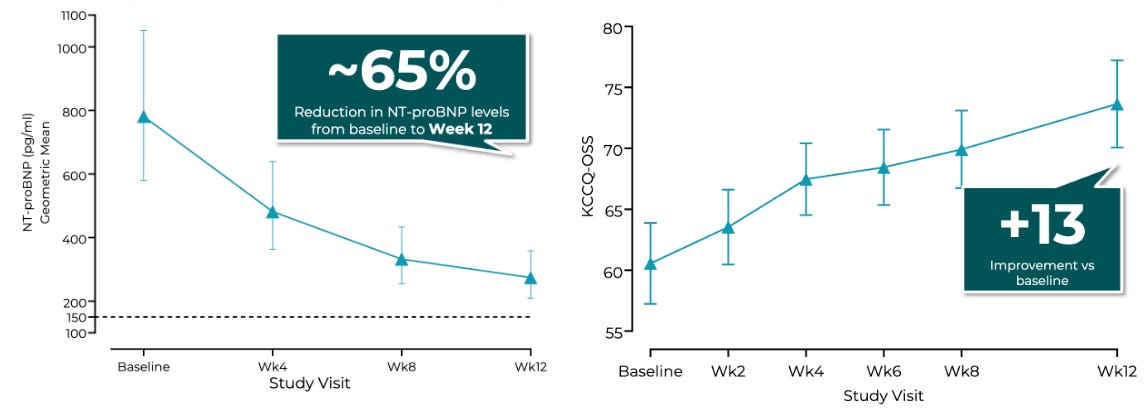
The multi-center, open-label, dose-finding Phase 2 CIRRUS-HCM trial evaluated flexible once-daily oral doses of EDG-7500 (25–150 mg) over 12 weeks in 53 symptomatic patients split across parallel obstructive (oHCM, N=20) and nonobstructive (nHCM, N=33) cohorts, utilizing titrated dosing guided by LVOT gradient reductions or NT-proBNP levels, respectively. The trial achieved encouraging efficacy: 74% of oHCM patients and 88% of nHCM patients achieved NT-proBNP biomarker normalization (<150 pg/mL) or a ≥50% reduction from baseline, alongside a mean 65% NT-proBNP decline in the nHCM group. Notably, nHCM patients demonstrated a mean 13-point improvement in KCCQ-OSS, numerically outperforming the ~10-point KCCQ-CSS improvement observed with Cytokinetics’ aficamten in the Phase 3 ACACIA-HCM trial. On safety, across over 700 serial echocardiograms, there was no mathematical relationship between drug exposure and LVEF reductions, with zero patients dropping below the critical 50% LVEF threshold.
Following the conclusion of Part D, Edgewise is utilizing the PK/PD data to finalize its registrational dose cohorts. Armed with cash reserves bolstered by a recent $2.65 billion asset sale to Servier, Edgewise plans to initiate its global Phase 3 pivotal trials for EDG-7500 in late 2026. Patients completing the 12-week CIRRUS-HCM study will continue tracking in an open-label extension for up to 18 months to gather long-term durability and safety metrics.
Sources: Edgewise press release, Edgewise slide deck, Weekly Readout on Phase 3 ACACIA-HCM trial for Cytokinetics’ aficamten
First approved PROTAC, obesity drug for alcoholism, and more

Link to Cytokinetics / Aficamten (cardiac myosin inhibitor) / Phase 3 (nHCM)
Love biotech? Check out Biotech Readout’s full content library, or navigate directly to a segment that interests you:
Frontiers in Medicine: Exploring the frontiers of our understanding and treatments for disease.
Medical History: Recovering forgotten relics in the history of medicine.
Acquisitions: Exploring the innovation behind acquired companies.
Weekly Readout: A digest of new clinical data from the past week.

To contact us, please send us an email at biotechreadout@gmail.com
Disclaimers
Investigational Status Disclaimer
The therapeutic candidates discussed in this newsletter are currently in clinical development and have not been approved for commercial sale by the U.S. Food and Drug Administration (FDA), the European Medicines Agency (EMA), or other global regulatory authorities. Their safety and efficacy have not been established. References to pipeline products and ongoing clinical trials involve significant risks and uncertainties. Statements regarding the potential safety, potency, or efficacy of investigational drugs reflect current hypotheses and are not a guarantee of future performance or regulatory clearance. The outcome of clinical trials is inherently unpredictable, and clinical results from earlier stages may not be predictive of results in later, larger-scale trials.
No Medical Advice Disclaimer
This newsletter is for informational and educational purposes only. The content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or another qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read in this publication.
No Patient-Provider Relationship Disclaimer
The information provided in this newsletter is for educational and analytical purposes only. Receipt of this information, or any interaction with this content, does not create a physician-patient, pharmacist-patient, or any other professional-provider relationship between you and the authors or publishers. This newsletter should not be used as a substitute for a personal consultation with a qualified healthcare professional.
Forward-Looking Statements Disclaimer
This newsletter contains “forward-looking statements” regarding future events, including clinical trial timing, regulatory milestones, and projected market performance. These statements are based on current expectations and assumptions that are subject to significant risks and uncertainties. Actual results may differ materially from those expressed or implied. We undertake no obligation to update these statements as a result of new information or future developments.
Third-Party Links & Content Disclaimer
This newsletter contains links to third-party websites, including clinical trial registries and corporate presentations. Biotech Readout does not endorse, guarantee, or assume responsibility for the accuracy or reliability of any information offered by third-party providers.
Errors and Omissions Disclaimer
While we strive for technical accuracy, the information in this newsletter is provided on an “as is” basis with no guarantees of completeness, accuracy, or timeliness. Biotech Readout assumes no liability for any errors or omissions in the content of this publication.
Non-Endorsement Disclaimer
Any reference to specific commercial products, processes, or services by trade name, trademark, or manufacturer does not constitute or imply an endorsement or recommendation by the author. All trademarks are the property of their respective owners.
No Investment Advice Disclaimer
This newsletter is for informational purposes only and does not constitute financial, investment, or legal advice. The author is not a registered investment advisor. You should consult with a professional financial advisor before making any investment decisions. The biotechnology sector is highly volatile; past performance is not indicative of future results.
Conflict of Interest Disclaimer
The author of this newsletter maintains a position of independence. At the time of publication, the author holds no direct financial interest, equity, or options in any of the companies mentioned in this report. No compensation has been received from any third party to feature or analyze specific therapeutic candidates or corporate entities.