
First approved PROTAC, obesity drug for alcoholism, and more
Weekly Readout #6: Week of May 4, 2026

Disclaimer: This newsletter is for educational and informational purposes only and does not constitute medical, investment, or financial advice, nor does it establish a provider-patient relationship. Content may include forward-looking statements and discussions of investigational therapeutic candidates that are not FDA/EMA approved; their safety and efficacy remain unestablished and clinical outcomes are unpredictable. While we strive for accuracy, all information is provided as is without guarantees. This newsletter is independent, and the author holds no financial positions in the companies mentioned nor receives third-party compensation for this coverage. Please find a complete version of our disclaimers at the bottom of this article and on our About page.

Press ⬇️ to go down to a section and ⬆️ to go back up to the Table of Contents
Table of Contents
This week, we discuss…
…landmark events:
First ever FDA-approved PROTAC, ushering in a brand new therapeutic modality; Arvinas & Pfizer / Veppanu (SERD) / FDA approval (2L ER+ HER2- ESR1m mBC) ⬇️
First randomized controlled trial testing obesity drug semaglutide in Alcohol Use Disorder; Investigator-sponsored / Semaglutide (GLP1 agonist) / Phase 2 (AUD) ⬇️
…potentially first-in-disease medicines:
In non-obstructive hypertrophic cardiomyopathy (nHCM); Cytokinetics / Aficamten (cardiac myosin inhibitor) / Phase 3 (nHCM) ⬇️
In a rare but devastating liver disease called primary sclerosing cholangitis (PSC); Mirum / Volixibat (iBAT inhibitor) / Phase 2b (PSC) ⬇️
…potentially best-in-disease medicines:
In second-line HR+ HER2- metastatic breast cancer regardless of PIK3CA mutation status; Celcuity / Gedatolisib (pan-PI3K/mTOR inhibitor) / P3 success (2L PIK3CAm HR+ HER2- mBC) ⬇️
In hidradenitis huppurativa (HS); Avalo / AVTX-009 (anti-IL1b mAb) / Phase 2 (HS) ⬇️
In inactive thyroid eye disease (TED); Viridian / Elegrobart (IGF-1R inhibitor) / Phase 3 (chronic/inactive TED) ⬇️
Arvinas & Pfizer / Veppanu (SERD) / FDA approval (2L ER+ HER2- ESR1m mBC)
On May 1, 2026, the FDA granted approval for Veppanu (vepdegestrant) for adults with ESR1-mutated, ER+/HER2- advanced or metastatic breast cancer who have progressed following at least one line of endocrine therapy. This is a landmark approval, as it represents the first-ever FDA-approved PROTAC (proteolysis-targeting chimera) therapy.
Indication: The pathophysiology of ESR1-mutated (ESR1m) metastatic breast cancer is driven by acquired ligand-independent activation of the estrogen receptor. While initial ER+ disease depends on circulating estrogen, chronic exposure to aromatase inhibitors (AIs) often triggers mutations in the ESR1 gene (most commonly in the Y537 or D538 residues of the ligand-binding domain). These mutations lock the estrogen receptor in a constitutively “on” conformation, allowing tumor cells to proliferate even when systemic estrogen levels are depleted. In the second-line (2L) setting, the standard of care has pivoted toward precision endocrine therapy following progression on a 1L CDK4/6 inhibitor plus an AI.
Mechanism: Veppanu is a heterobifunctional protein degrader that utilizes PROTAC technology to eliminate the estrogen receptor (ER) rather than just blocking it. It binds simultaneously to the ER and an E3 ubiquitin ligase. This tags the ER for destruction by the cell’s natural waste disposal system, the proteasome. Unlike traditional selective estrogen receptor degrader (SERDs) or aromatase inhibitors, Veppanu effectively degrades ESR1 mutations, which are common drivers of resistance (found in 40-50% of patients) after exposure to initial endocrine therapies.
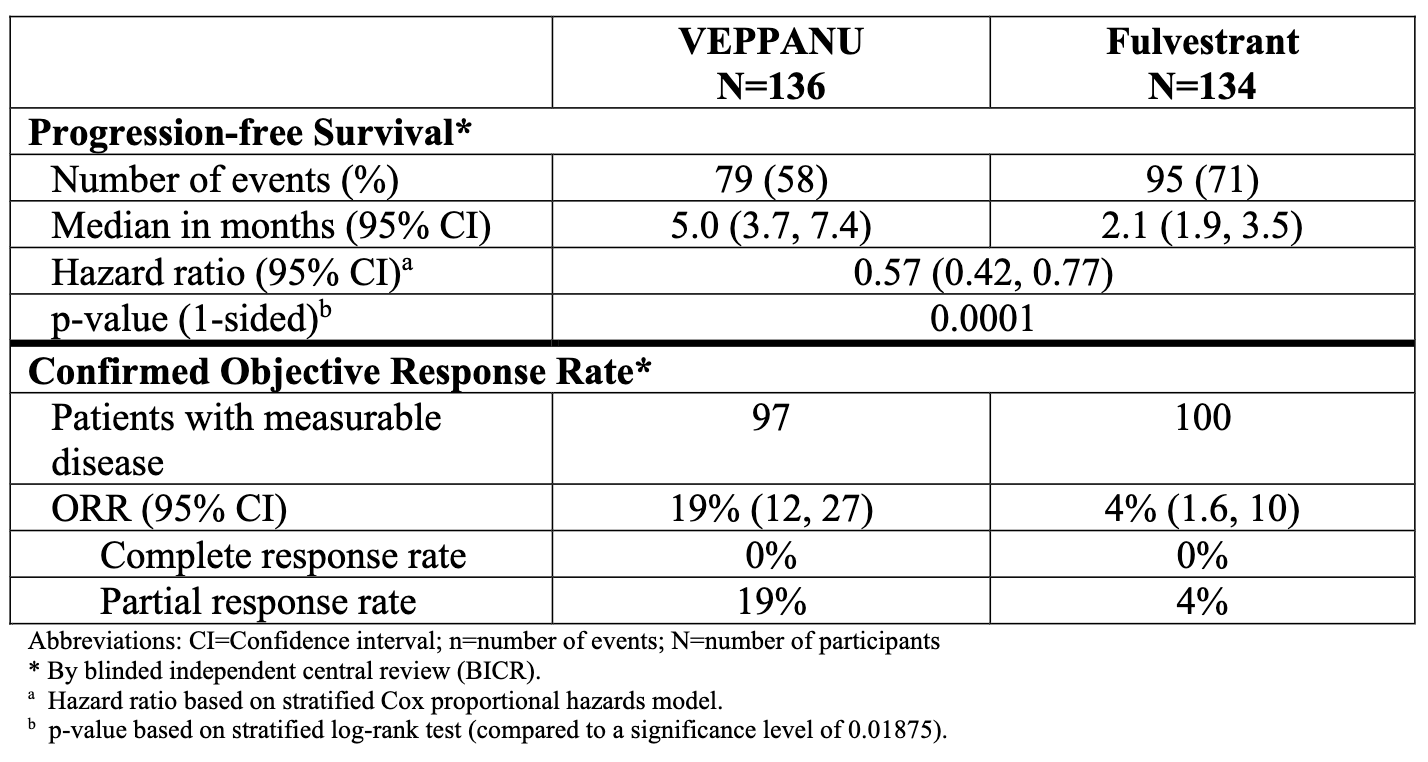
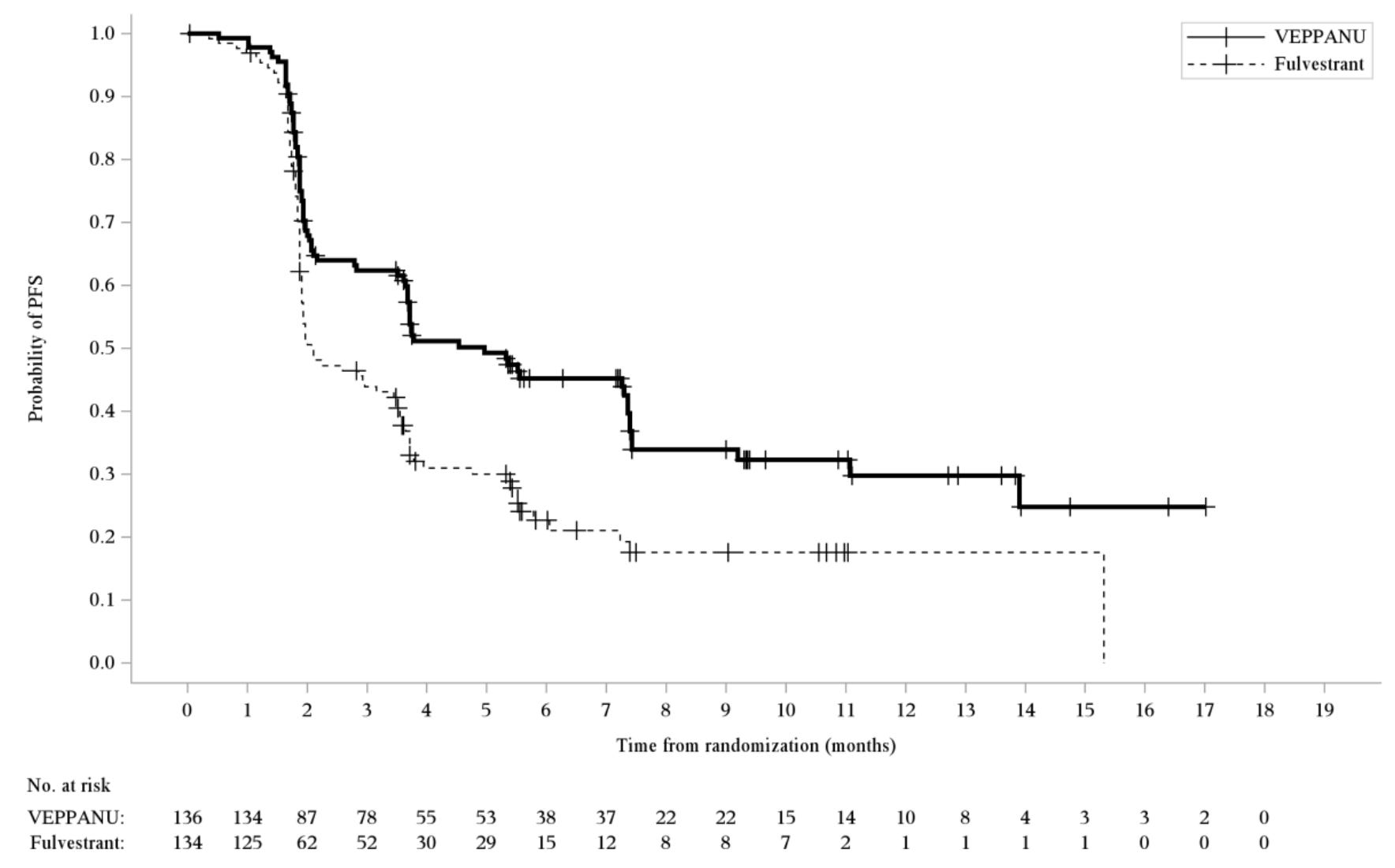
Trial design: The approval centered on the Phase 3 VERITAC-2 study in which 624 patients with ER+/HER2- advanced breast cancer (270 patients had confirmed ESR1 mutations; the group for which the drug was approved) who had progressed on 1–2 lines of endocrine therapy (including at least one line with a CDK4/6 inhibitor) were randomized 1:1 to receive either oral Veppanu (200 mg daily) or intramuscular Faslodex (current standard of care). The major efficacy outcome was progression-free survival (PFS) as assessed by blinded independent central review (BICR) in the population of patients whose tumors had an ESR1 mutation and in the overall population evaluated according to Response Evaluation Criteria in Solid Tumors (RECIST), version 1.1. Additional efficacy outcomes were overall survival (OS) and objective response rate (ORR) as assessed by BICR.
Data (press release, label): A statistically significant difference in PFS by BICR was observed for the patients whose tumors had ESR1 mutations for Veppanu compared with Faslodex/fulvestrant (see table and graph below). Overall survival was immature with 16% of deaths in this population at the time of the PFS analysis. Most common (≥10%) adverse reactions with Veppanu, includinglaboratory abnormalities, were decreased white blood cells, increased AST, musculoskeletal pain, fatigue, decreased hemoglobin, decreased neutrophils, increased ALT, increased alkaline phosphatase, nausea, decreased bloodpotassium, increased bilirubin, decreased appetite, electrocardiogram QT-prolonged, decreased platelets, and constipation. The label includes warnings for QTc interval prolongation and embryo-fetal toxicity.
Impact:
First-in-Modality: Validates PROTAC technology as a viable modality for cancer treatment.
Oral Convenience: Provides an oral alternative to the intramuscular injections required for fulvestrant.
Precision Medicine: The FDA simultaneously approved the Guardant360 CDx as a companion diagnostic to identify ESR1 mutations via ctDNA (blood test), cementing molecular profiling as a standard step in treatment selection.
Next steps: The trial sponsors (Arvinas and Pfizer) intend to jointly identify and select a third-party partner with the capabilities and expertise to maximize the commercial potential of Veppanu. The companies are on track to announce selection of a third party. Ongoing studies are evaluating vepdegestrant in combination with other agents (e.g., CDK4/6 inhibitors) to see if earlier lines of therapy can benefit. This success paves the way for the trial sponsor’s other PROTAC candidates targeting neurodegenerative and neuromuscular diseases.
Investigator-initiated / Semaglutide (GLP1 agonist) / Phase 2 (AUD)
On May 2, 2026, the Lancet published the first large-scale, double-blind, randomized controlled trial (RCT) specifically using semaglutide (the 2.4 mg Wegovy dose approved for obesity) to treat Alcohol Use Disorder (AUD). The study was led by Professor Anders Fink-Jensen and Dr. Mette Kruse Klausen at the Mental Health Center Copenhagen in Copenhagen, Denmark (Rigshospitalet). The trial was funded by the Novo Nordisk Foundation, which is the controlling shareholder of Novo Nordisk, the manufacturer of Wegovy (semaglutide). However, the study was truly “investigator-initiated,” meaning the foundation provided the funding as a grant, but the company (Novo Nordisk) was not involved in the study’s design, execution, or data analysis.
Note that semaglutide is not FDA-approved to treat alcohol use disorder and Novo Nordisk is “not currently conducting any dedicated clinical studies to evaluate marketed semaglutide products … in patients with substance use disorders or addiction-related illnesses”, according to the company. As of May 2026, semaglutide remains only FDA-approved for Type 2 Diabetes (Ozempic/Rybelsus) and Chronic Weight Management (Wegovy). Neither the author nor this publication encourages or condones the off-label use of semaglutide.
Indication: Alcohol Use Disorder (AUD) is characterized by a dysregulation of the brain’s reward and executive control systems, primarily involving the chronic adaptation of GABAergic (inhibitory) and glutamatergic (excitatory) neurotransmission. Prolonged alcohol consumption downregulates GABA receptors and upregulates NMDA receptors, leading to a state of hyperexcitability during withdrawal that drives compulsive seeking behavior through the mesolimbic dopamine pathway. The current standard of care employs a multimodal approach, combining psychosocial interventions—such as Cognitive Behavioral Therapy (CBT), Motivational Enhancement Therapy (MET), and 12-step programs with FDA-approved pharmacotherapies. These can include naltrexone (an opioid antagonist to reduce cravings), acamprosate (to stabilize glutamate signaling), and disulfiram (an aversive agent that induces illness upon alcohol ingestion), though clinical uptake of these medications remains low despite their efficacy.
Mechanism: Semaglutide is a GLP-1 receptor agonist (GLP-1RA). While its primary metabolic function is increasing insulin secretion and slowing gastric emptying (we covered this in our “Frontiers in Medicine” piece on Obesity, Part 2), its effect on addiction is centered in the central nervous system. It is believed to modulate the brain’s reward system, specifically the mesolimbic dopamine pathway (the ventral striatum). By interacting with these receptors, semaglutide reduces the dopamine rush associated with alcohol consumption, effectively dampening the reinforcing pleasure of drinking and reducing the psychological urge or craving. Nevertheless, this mechanism is largely theoretical.
Trial design: The Phase 2 was a randomized, double-blind, placebo-controlled trial evaluating once-weekly subcutaneous semaglutide (escalated to 2.4 mg) versus placebo over 26 weeks in 108 adults with comorbid obesity (BMI ≥30) and Alcohol Use Disorder (AUD) seeking treatment. The primary endpoint was the change in the number of heavy drinking days (HDD) from baseline to week 26.
Data (paper): The trial demonstrated a significant effect in reducing alcohol consumption. Semaglutide recipients saw a -41.1% reduction in heavy drinking days compared to -26.4% in the placebo group at week 26. he semaglutide group reduced their monthly intake by ~1,550g (compared to ~1,026g for placebo). As the investigators expected, the semaglutide group lost significantly more weight (~11.2 kg vs ~2.2 kg). The profile was consistent with other GLP-1 trials; gastrointestinal issues (nausea, vomiting) were the most common adverse events.
Impact:
Magnitude of Effect: The reduction in WHO drinking-risk levels was notably higher than standard-of-care AUD medications like naltrexone or acamprosate. The publication specifically highlights a massive potential benefit for the estimated 8 million Americans who suffer from both obesity and AUD. However, semaglutide would have to be investigated in manufacture-sponsored trials and reviewed by regulators for this to come to fruition.
Validation of Anecdotes: This trial provides the first high-quality clinical evidence to back up years of anecdotal reports from Wegovy users claiming they lost interest in alcohol.
Next steps: Despite the clinical success of this investigator-initiated trial, the path to formal FDA approval for semaglutide in AUD is currently stagnant. The company has explicitly stated they are not currently conducting dedicated studies for AUD. Their focus remains on diabetes, obesity, and cardiovascular/kidney outcomes. Since this trial focused on patients with obesity, more research is needed to determine if the same effect occurs in lean patients with AUD. The academic researchers who conducted this trial called for larger, multi-center Phase 3 trials and longer follow-up periods to see if the “sobriety” effect persists after the drug is discontinued. Notably, Eli Lilly is evaluating a GLP1/GIP dual agonist called brenipatide for alcohol use disorder (AUD) in two Phase 3 clinical trials with estimated completion dates in April 2028.
Cytokinetics / Aficamten (cardiac myosin inhibitor) / Phase 3 (nHCM)
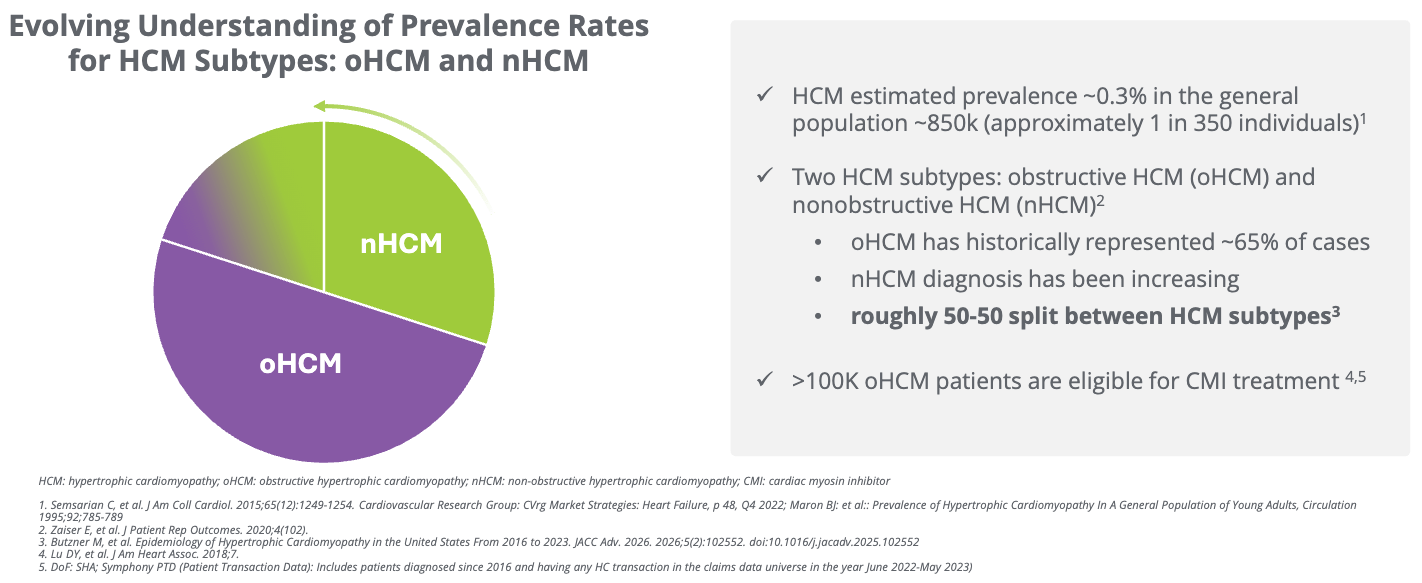
The Phase 3 ACACIA-HCM trial results for aficamten in non-obstructive hypertrophic cardiomyopathy (nHCM) represent a major milestone, as this is the first cardiac myosin inhibitor to succeed in this specific, hard-to-treat patient population. Last year, aficamten was approved under the trade name Myqorzo by the FDA to treat adults with symptomatic obstructive hypertrophic cardiomyopathy (oHCM). This Phase 3 data could support a future FDA approval, pending further regulatory review, roughly doubling the number of patients that Myqorzo could treat, according to Cytokinetics’ estimates (see the figure below).
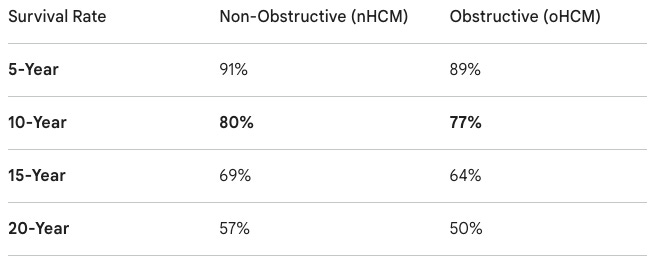
Indication: The pathophysiology of non-obstructive hypertrophic cardiomyopathy (nHCM) is characterized by genetic mutations in the cardiac sarcomere that lead to excessive heart muscle thickening (hypertrophy) without a physical blockage in the left ventricular outflow tract. This results in diastolic dysfunction, where the thickened ventricle becomes stiff and fails to fill properly, alongside hyperdynamic contractility that increases myocardial oxygen demand. Over time, this cellular stress leads to interstitial fibrosis and elevated filling pressures, causing symptoms like exertional dyspnea, fatigue, and chest pain. Historically, the standard of care has been limited to symptom management using non-specific medications such as beta-blockers or calcium channel blockers to slow the heart rate and improve diastolic filling; however, these do not address the underlying disease mechanism, leaving a significant unmet need for targeted therapies. A retrospective study led by Dr. Milind Desai at the the Cleveland Clinic following 3,393 patients (Jadam et al. JACC (2025)) found that 20-year survival was as low as 57% for nHCM and 50% for oHCM. While nHCM carries a slightly better survival profile than oHCM, the high rate of arrhythmic risk and the lack of targeted therapies have made it a particularly challenging disease for cardiologists to manage.
Mechanism: Aficamten is a next-generation cardiac myosin inhibitor. It binds selectively to cardiac myosin, reducing the number of active myosin heads that can bind to actin. By dampening hypercontractility (a hallmark of HCM), it reduces the excessive force of heart muscle contraction. In nHCM, where there is no physical blockage (outflow tract obstruction), the drug works by improving diastolic relaxation and lowering the metabolic stress on the heart muscle.
Trial design: The Phase 3 ACACIA-HCM trial was a double-blind, randomized, placebo-controlled trial comparing aficamten versus placebo for 36 weeks with a dose-titration period guided by echocardiography (ECG) in adults with symptomatic non-obstructive HCM (nHCM). The trial had a dual primary endpoint, meaning that it needed to show a statistically significant improvement in at least one of these endpoints to be deemed a “success”. They included:
Change in KCCQ-CSS (Kansas City Cardiomyopathy Questionnaire - Clinical Summary Score) at Week 36.
Change in pVO2 (Peak Oxygen Consumption during exercise) at Week 36.
Data (press release): The trial met both dual primary endpoints with statistical significance (see the graphs below). Additionally, key secondary endpoints were met with high significance (p < 0.001), including improvements in NYHA Functional Class and reduction in NT-proBNP (a marker of cardiac wall stress), although numerical values were not shared in this release. There were “no new safety signals” associated with aficamten, according to the trial sponsor. Left ventricular ejection fraction (LVEF) < 50% occurred in 10% of aficamten patients vs. 1% in placebo, with most cases being asymptomatic and reversible upon dose adjustment or interruption. This is important because if LVEF drops too low, it can lead to systolic heart failure. The label requires clinicians to pause or “ct” (continue/titrate) therapy if LVEF falls below 50%, as this indicates the heart is no longer pumping effectively enough to meet the body’s needs. The study data showed that most LVEF drops were asymptomatic and resolved once the drug was paused, suggesting that the “stop-start” guidance on the label is a potentially effective mechanism for managing the drug’s primary effect.
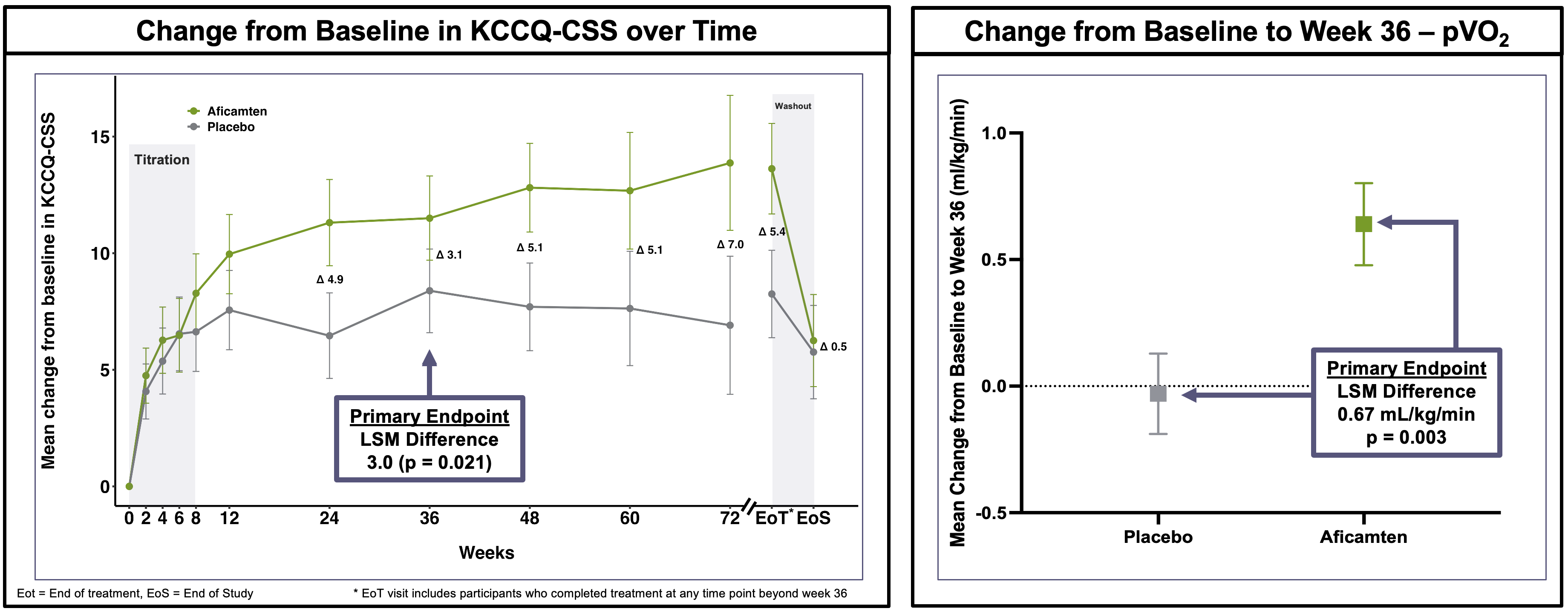
Impact:
First-in-Indication Potential: While aficamten is already FDA-approved for obstructive HCM (oHCM), this Phase 3 data could support a future FDA approval in non-obstructive hypertrophic cardiomyopathy (nHCM), pending further regulatory review.
Patient Benefit: The data shows patients feel better (KCCQ) and can physically do more (pVO2), addressing the high unmet need for the roughly 30% of HCM patients who do not have an obstruction.
Competitive Edge: This readout is particularly significant because the competitor drug, mavacamten (approved for oHCM as Camzyos), previously failed to show significant benefit in its own Phase 3 nHCM trial. This cements aficamten as the leader in this segment.
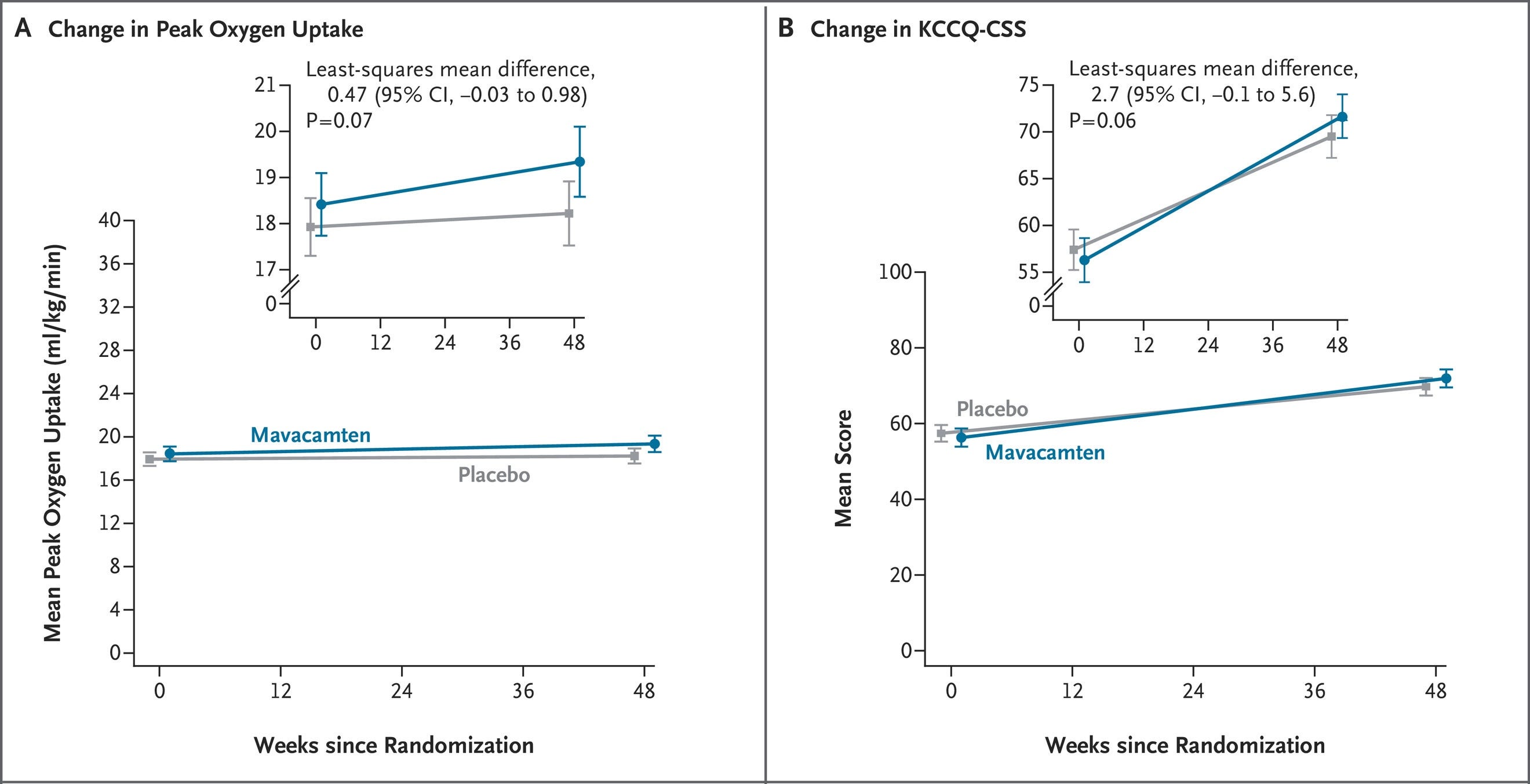
Next steps: Full data results will be presented at an upcoming major medical meeting (possibly ESC Heart Failure or similar). Cytokinetics has 10 presentations scheduled for the European Society of Cardiology (ESC) Heart Failure Congress in Barcelona (May 9–12, 2026). This includes a Late-Breaking Science presentation on the dose-dependent effects of aficamten (from the MAPLE-HCM study). The trial sponsor plans to “discussing [these results] with the U.S. FDA and other regulatory authorities” for the nHCM indication.
Mirum / Volixibat (iBAT inhibitor) / Phase 2b (PSC)
Mirum Pharmaceuticals announced positive topline results from its VISTAS Phase 2b study of volixibat on May 4, 2026. This data de-risks the asset for Primary Sclerosing Cholangitis (PSC), a rare liver disease with no currently approved treatments for cholestatic pruritus (itch).
Indication: Primary Sclerosing Cholangitis (PSC) is a chronic, progressive liver disease characterized by immune-mediated inflammation, thickening, and obliterative fibrosis of both intrahepatic and extrahepatic bile ducts. This beading of the ducts leads to cholestasis (impeded bile flow), which eventually causes biliary cirrhosis, portal hypertension, and an increased risk of cholangiocarcinoma. While the exact etiology remains unknown, the strong association with inflammatory bowel disease (IBD), present in roughly 70–80% of patients, suggests a complex interplay between genetics, gut microbiota, and immune dysregulation. Currently, there is no FDA-approved treatment that halts disease progression. Management focuses on symptom relief, such as using ursodeoxycholic acid (UDCA) for biochemical improvement (though its impact on survival is debated), endoscopic balloon dilation for dominant strictures, and ultimately, liver transplantation for end-stage disease. PSC patients have a three- to four-fold increased risk of all-cause mortality compared to the general population. The median time from diagnosis to either liver transplantation or death is approximately 15 to 21 years. This range varies significantly; for patients seen at major transplant centers (who often have more advanced disease), the median survival may be as low as 13 years. For those who undergo a liver transplant, the prognosis is generally good, with a 5-year survival rate of approximately 85%.
Mechanism: Volixibat is an oral, minimally absorbed, potent ileal bile acid transporter (IBAT) inhibitor. It blocks the apical sodium-dependent bile acid transporter (ASBT/SLC10A2) in the terminal ileum. By preventing the reabsorption of bile acids (which are normally 95% recycled), it promotes their fecal excretion. Reducing the systemic and hepatic bile acid pool is thought to lower the concentration of pruritogens (itch-inducing substances) in the blood, thereby alleviating severe cholestatic pruritus.
Trial design: The VISTAS Phase 2b trial evaluated volixibat 20 mg twice daily (BID) versus placebo in 158 patients with PSC and cholestatic pruritus (111 primary analysis cohort had moderate-to-severe itch, 47 secondary analysis cohort had mild itch). The primary endpoint was mean change in weekly averaged daily itch score (Adult ItchRO scale, 0–10) from baseline to the average of the last 12 weeks of a 28-week treatment period.

Data (press release, slide deck): The study met its primary endpoint with high statistical significance. It also met key secondary endpoints, showing more patients with a ≥2 point reduction in Adult ItchRO (55.6% volixibat versus 26.3% placebo, p=0.0019) and a greater reduction in serum bile acids (-33 volixibat versus +2.1 placebo, p=0.0019; side-note: they didn’t include units in the press release). Statistically significant reductions in pruritus were observed as early as Week 2. The profile was “consistent with the IBAT inhibitor class” according to the trial sponsor, with higher incidence of diarrhea (40.3% volixibat versus 8.6% placebo) and numerically higher discontinuations due to AEs (9.1% volixibat versus 2.5% placebo).
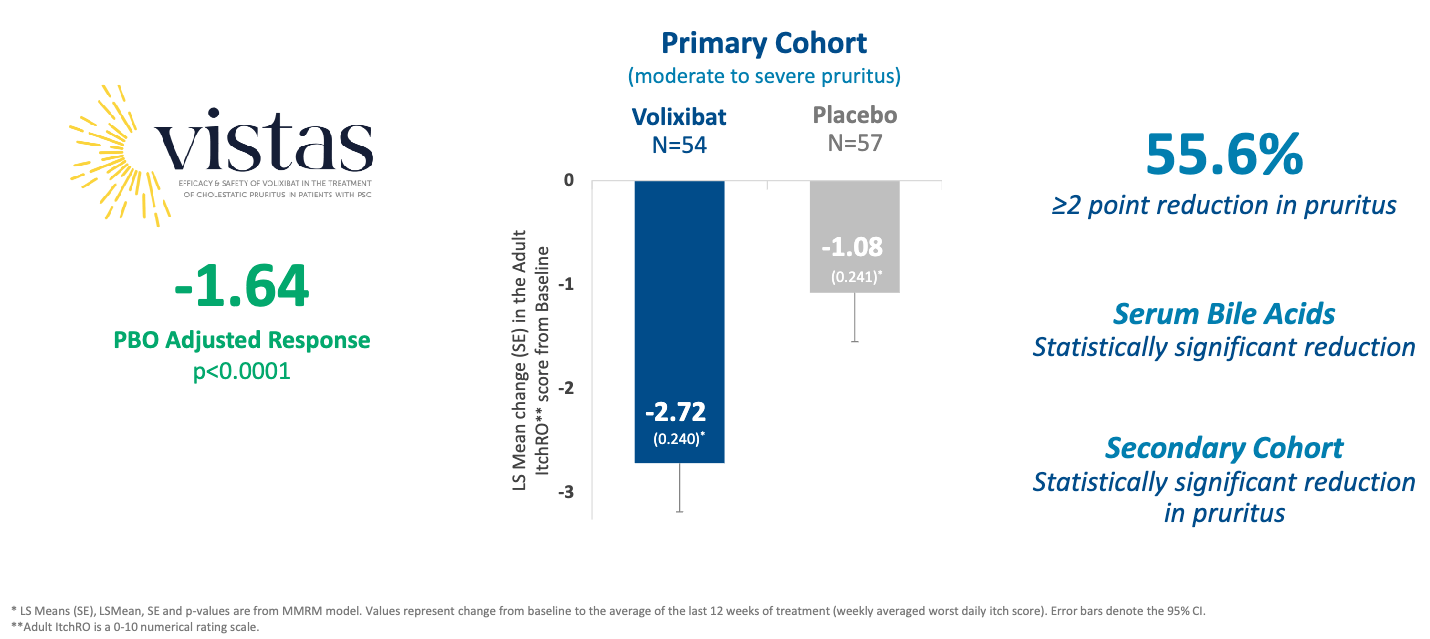
Impact:
Potentially first-in-indication: There are currently no FDA-approved therapies specifically for the treatment of pruritus in PSC. Volixibat is now positioned to potentially be the first.
Quality of Life: The ~2.7 point reduction is considered clinically meaningful, as itch in PSC is often debilitating, leading to sleep deprivation and significant psychological distress.
Class Validation: This success further validates Mirum’s IBAT platform, which already includes Livmarli (maralixibat) for Alagille syndrome and PFIC.
Next steps: The trial sponsor plans to present this data as a late-breaking oral presentation at the EASL Congress on May 30, 2026. They also anticipate a pre-NDA meeting with the FDA, scheduled for Summer 2026, with an NDA submission in 2H 2026. The trial sponsor is also evaluating volixibat in the VANTAGE Phase 2b study for Primary Biliary Cholangitis (PBC), with topline data expected in Q1 2027.
Celcuity / Gedatolisib (pan-PI3K/mTOR inhibitor) / P3 success (2L PIK3CAm HR+ HER2- mBC)
On May 1, 2026, Celcuity announced positive topline Phase 3 results for the PIK3CA-mutated cohort of the VIKTORIA-1 trial. This follows the company’s ongoing regulatory momentum, with a PDUFA date for the PIK3CA wild-type population set for July 17, 2026. This is a descriptive data release without numerical data, but it’s important enough that it merits its own section.

Indication: The pathophysiology of PIK3CA-mutated (PIK3CAm) HR+/HER2- metastatic breast cancer is characterized by the hyperactivation of the PI3K/AKT/mTOR pathway, which serves as a critical escape mechanism for tumor cells to bypass estrogen receptor (ER) inhibition. Found in approximately 40% of cases, PIK3CA mutations (most commonly in the H1047R, E542K, or E545K domains) lead to constitutive signaling of the p110α catalytic subunit, driving cell proliferation and survival even under the pressure of endocrine therapy. In the second-line (2L) setting, the established standard of care for these patients is Piqray (alpelisib), a p110α-selective PI3K inhibitor, in combination with Faslodex (fulvestrant), which significantly improved progression-free survival (PFS) in the SOLAR-1 trial. Emerging therapies like gedatolisib (a pan-PI3K/mTOR inhibitor) are actively shifting the benchmark toward triplet combinations or more potent, selective inhibition to overcome the resistance mechanisms associated with first-generation PI3K inhibitors.
Mechanism: Gedatolisib is a highly potent, pan-class I PI3K and mTORC1/C2 inhibitor. It targets the interconnected PI3K/AKT/mTOR (PAM) pathway at multiple points. Unlike single-target inhibitors (e.g., Piqray/alpelisib, which targets only PI3K-alpha, gedatolisib inhibits all four PI3K isoforms plus both mTOR complexes. By blocking both upstream and downstream components, gedatolisib prevents the adaptive resistance (cross-activation of PI3K isoforms) that typically occurs when a tumor is treated with an isoform-specific inhibitor. In the triplet regimen, it works alongside estrogen receptor degrader therapy (Faslodex/fulvestrant) and a CDK4/6 inhibitor (Ibrance/palbociclib) to simultaneously shut down the three primary drivers of ER+ breast cancer growth.
Trial design: The Phase 3 VIKTORIA-1 evaluated HR+/HER2- advanced breast cancer who had progressed on a prior CDK4/6 inhibitor plus an aromatase inhibitor. The latest data release focuses on “Study 2”, a cohort of patients who have PIK3CA mutations. It tested three arms:
Data (press release): The topline results demonstrated that both gedatolisib-based regimens (Arm 1 Triplet and Arm 2 Doublet) were superior to the current SOC (Piqray/Faslodex) at improving progression free survival (PFS) with statistical significance. Detailed figures will be presented at 2026 ASCO Annual Meeting, which is scheduled to take place from May 29 to June 2, 2026.
Impact:
Potentially New Standard-of-Care: By beating Piqray head-to-head, gedatolisib establishes itself as the potential new “best-in-class” PI3K-pathway inhibitor for 2L PIK3CAm HR+ HER2- mBC. According to the trial sponsor’s estimates, HR+/HER2- breast cancer is the most common subtype of breast cancer (70% of all breast cancers), of which approximately 40% have PIK3CA mutations.
Overcoming Resistance: The data suggests that gedatolisib can resensitize tumors to CDK4/6 inhibition (palbociclib) even after they have already progressed on that class of therapy.
Pan-Population Efficacy: Unlike many competitors that only work in mutated populations, gedatolisib has now shown Phase 3 success in both PIK3CA-mutated (this release) and PIK3CA-wild-type patients (earlier release, data shown in the graph below).
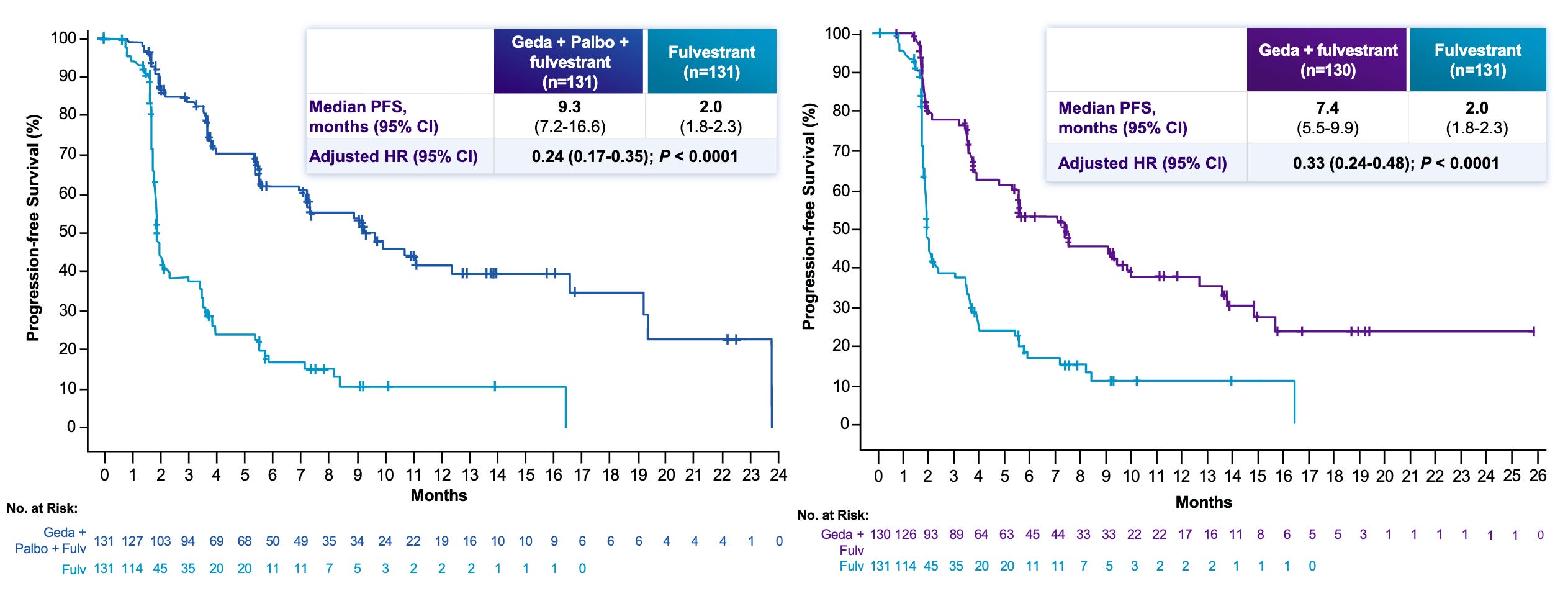
Next steps: The trial sponsor plans to present full data, including specific median PFS months and Hazard Ratios (HR), will be shared during a late-breaking oral session at ASCO 2026. They also intend to file a Supplemental New Drug Application (sNDA) for the PIK3CA-mutated population based on these results. The FDA is currently reviewing the first NDA for gedatolisib in the PIK3CA wild-type population under Priority Review (July 17, 2026 PDUFA). The trial sponsor is already enrolling patients in a Phase 3 trial evaluating gedatolisib in the first-line setting (treatment-naïve) to move the therapy earlier in the treatment paradigm.
Avalo / Abdakibart (anti-IL1b mAb) / Phase 2 (HS)
Avalo Therapeutics announced positive topline results for its Phase 2 LOTUS trial evaluating abdakibart (AVTX-009) in moderate to severe Hidradenitis Suppurativa (HS).
Indication: Hidradenitis Suppurativa (HS) is a chronic inflammatory skin disease primarily driven by the occlusion of the hair follicle (infundibulum), leading to follicular rupture and the subsequent release of keratin and bacteria into the dermis. This triggers a potent immune response involving the overexpression of cytokines like TNF-α, IL-17, and IL-1β, resulting in painful abscesses, nodules, and the formation of extensive scarring and draining tunnels (sinus tracts). The standard of care follows a staged approach: mild cases are managed with topical antibiotics (clindamycin) or resorcinol, while moderate-to-severe disease requires systemic intervention. This typically begins with oral antibiotics (tetracyclines or clindamycin/rifampin) as first-line therapy, followed by biologics, most notably TNF-inhibitors like Humira (adalimumab) or IL-17A inhibitors like Cosentyx (secukinumab), with surgical debridement or “unroofing” reserved for treating persistent tunnels and refractory lesions.
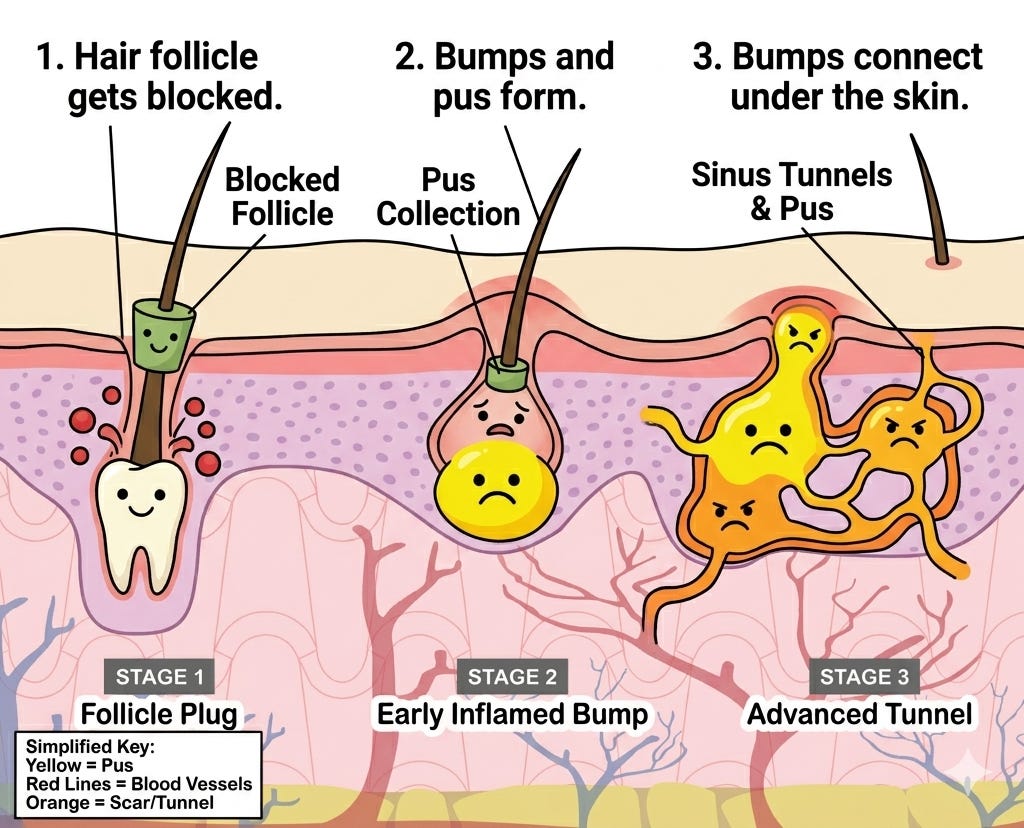
Mechanism: Abdakibart is a humanized monoclonal antibody (IgG4) that targets and inhibits interleukin-1 beta (IL-1β) signaling. IL-1β is a central driver of the inflammatory cascade in HS, contributing to the formation of painful nodules, abscesses, and draining tunnels. By neutralizing IL-1β, abdakibart aims to reduce systemic and local inflammation.
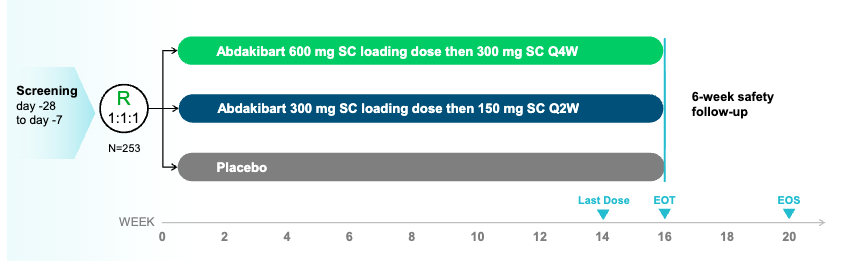
Trial design: The Phase 2 LOTUS trial was a randomized, double-blind, placebo-controlled trial evaluating two dosing schemes of abdakibart (600mg loading then 300 mg every 4 weeks (Q4W); 300mg loading then 150 mg every 2 weeks (Q2W)) against placebo in 253 adults with moderate to severe HS. The primary endpoint was the proportion of subjects achieving HiSCR75 (75% reduction in total abscess and inflammatory nodule [AN] count) at Week 16.
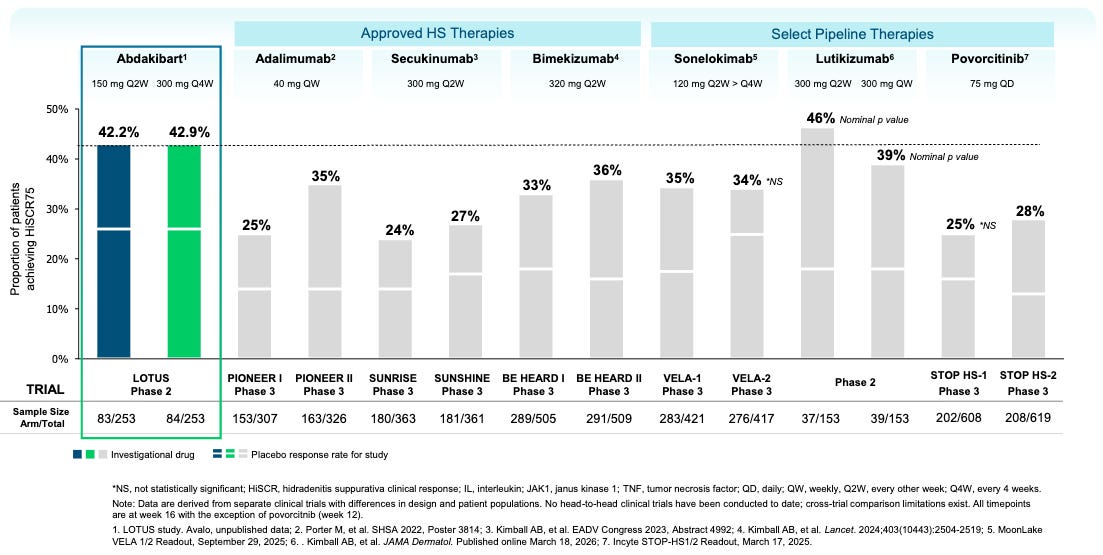
Data (press release, slide deck): The trial met its primary endpoint with statistical significance for both dose regimens (16-week HiSCR75 rates of 42.2% and 42.9% for Q2W and Q4W doses respectively versus 25.6% placebo, p=0.018 and 0.015 respectively). Based on the cross-trial comparison provided in the LOTUS study topline results, abdakibart demonstrates numerically competitive HiSCR75 rates compared to both approved therapies and other prominent pipeline candidates (see below). The caveat is that these data are derived from separate clinical trials with differences in design and patient populations. No head-to-head clinical trials have been conducted to date. All timepoints shown in the figure are at Week 16, with the exception of povorcitinib, which is at Week 12.
Impact:
Potentially Best-in-Disease: The 42.9% HiSCR75 rate is among the highest observed in a trial of this size for HS. A caveat is that, on a placebo-adjusted basis, abdakibart’s HiSCR75 rate comes in numerically lower than competing anti-IL-1β antibody lutikizumab (~17% abda versus ~28% lutiki). Given very high placebo variability across trials (ranging from 13% to 26% in the abdakibart trial), the most precise statement that we can make at this point is that abdakibart is roughly “on par” with competitive molecules. Only a large head-to-head trial would be able to infer non-inferiority/superiority with high precision.
Dosing Convenience: The success of the 300 mg every-4-weeks (E4W) regimen provides a patient-friendly monthly dosing option, which is a competitive advantage over more frequent treatments.
Target Validation: Reinforces the critical role of IL-1β in HS pathology, potentially positioning it as a preferred pathway over IL-17 or TNF for certain patient subsets.
Next steps: The trial sponsor plans to present detailed results at an upcoming medical conference (likely a major dermatology congress). They have also confirmed plans to advance abdakibart into a registrational Phase 3 program for HS. The company is exploring abdakibart in other immune-mediated inflammatory diseases.
Viridian / Elegrobart (IGF-1R inhibitor) / Phase 3 (chronic/inactive TED)
Viridian Therapeutics recently announced positive topline results for elegrobart (VRDN-003) from its Phase 3 REVEAL-2 trial in patients with chronic/inactive Thyroid Eye Disease (TED). This readout follows the REVEAL-1 success in active TED, positioning elegrobart as a potential best-in-class subcutaneous (SC) treatment.
Indication: Chronic (inactive) Thyroid Eye Disease (TED) is a late-stage autoimmune condition where the initial inflammatory “hot” phase has subsided, but the underlying tissue remodeling has left permanent structural damage. The pathophysiology centers on the activation of orbital fibroblasts via the IGF-1R and TSHR signaling complexes; in the chronic phase, this leads to significant fibrosis (scarring) and adipogenesis (fat expansion) within the confined orbital space. This volume increase results in persistent proptosis (bulging eyes), restricted extraocular muscle movement (leading to double vision), and potential optic nerve compression. Because inflammation is no longer the driver, the standard of care traditionally shifts from immunosuppression to mechanical intervention. Management typically involves rehabilitative surgeries performed in a specific sequence: orbital decompression to reduce pressure, followed by strabismus surgery to align the eyes, and finally eyelid retraction repair. However, the recent emergence of IGF-1R inhibitors like Tepezza (teprotumumab), and potentially elegrobart, has begun to shift the paradigm by offering a non-surgical medical option to reduce proptosis even in this inactive, fibrotic stage.
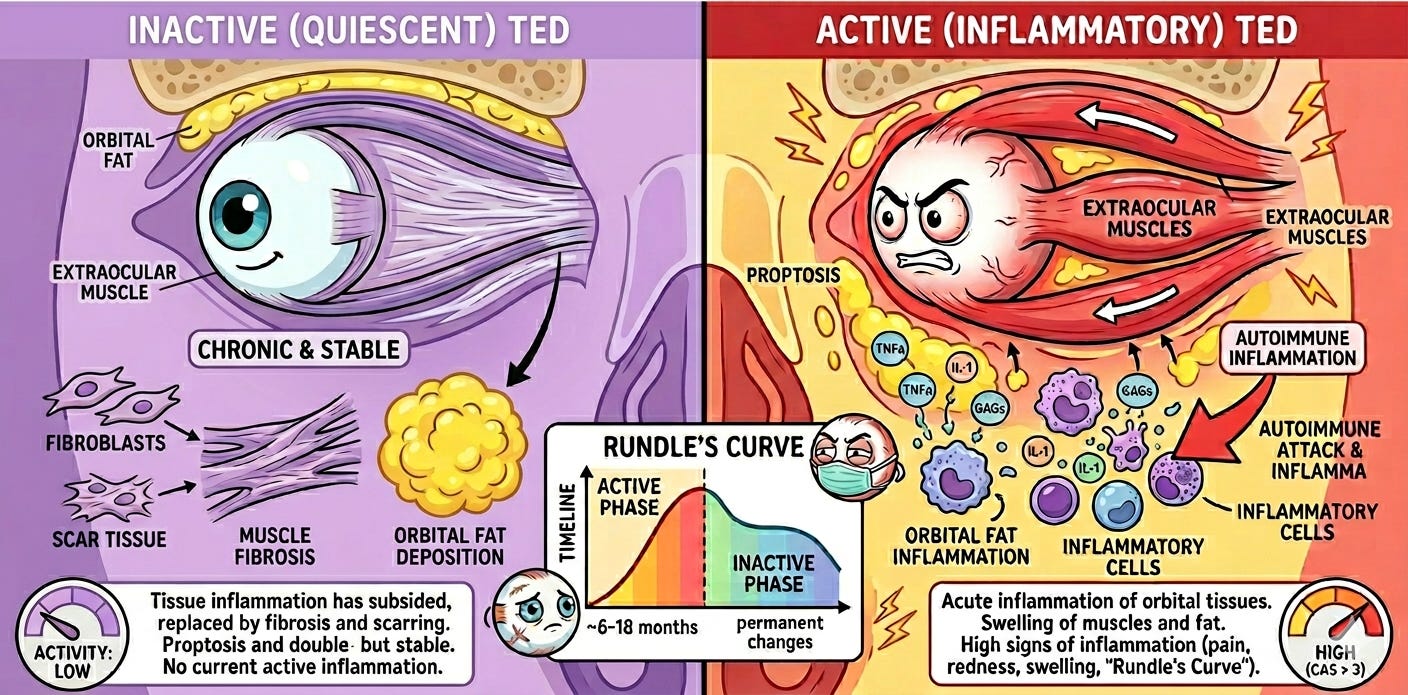
Mechanism: Elegrobart is an anti-Insulin-like Growth Factor-1 Receptor (IGF-1R) monoclonal antibody delivered subcutaneously via a low-volume autoinjector. It is a half-life extended version of veligrotug (VRDN-001). This allows for less frequent dosing (Q4W or Q8W) and home administration, compared to the intravenous (IV) infusions required for the current market leader, Tepezza.
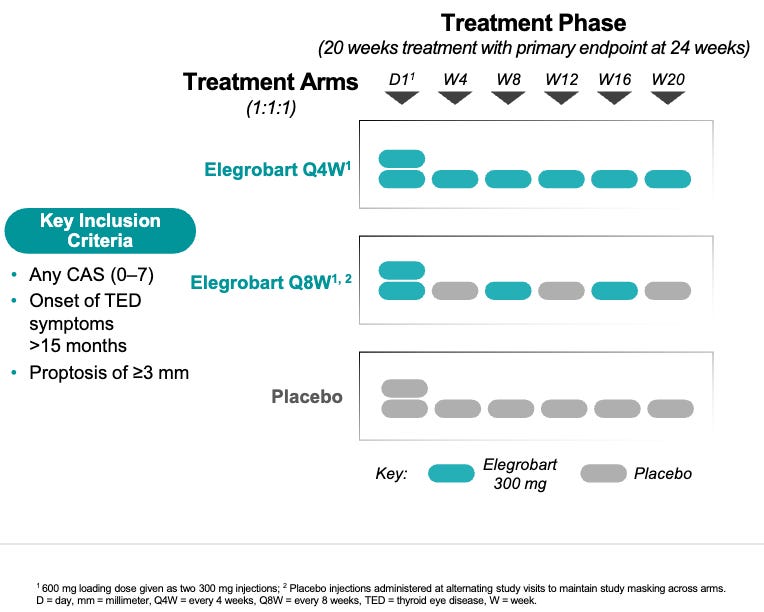
Trial design: The Phase 3 REVEAL-2 evaluated two dose schedules of elegrobart (Q4W/once every 4 weeks and Q8W/once every 8 weeks) against placebo in 204 patients with chronic/inactive TED (characterized by lower inflammation/CAS but persistent proptosis). The primary endpoint was the proptosis responder rate (reduction of ≥2mm) at Week 24.
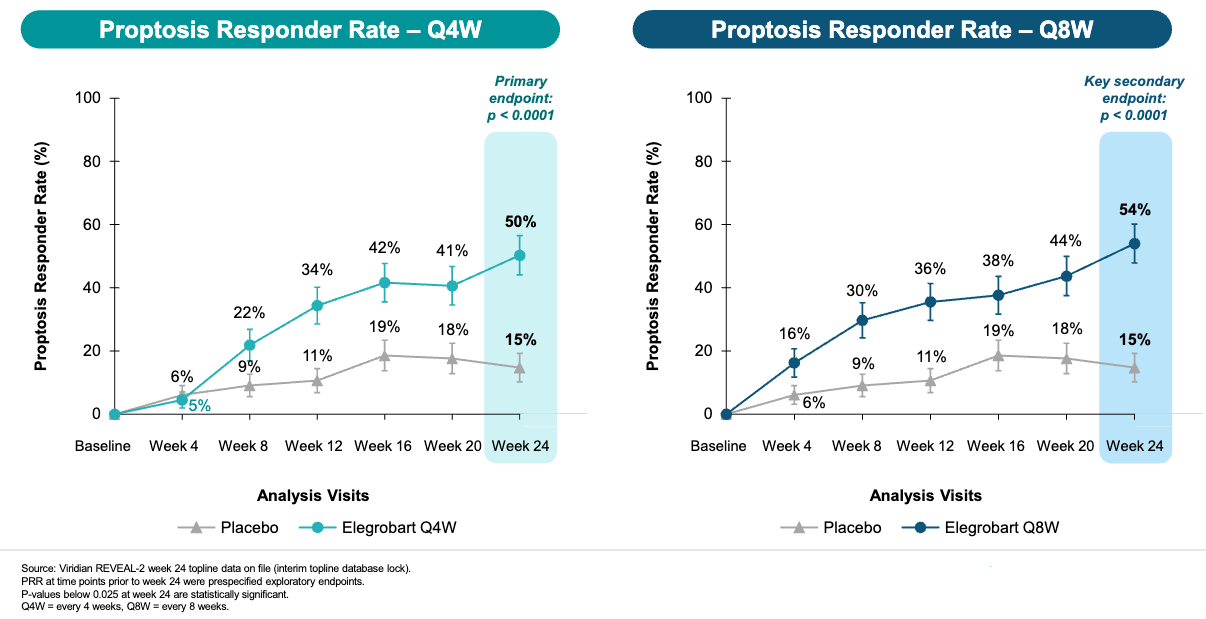
Data (press release, slide deck): Both doses met the primary endpoint with high statistical significance (see graphs below). Elegrobart also became the first subcutaneous (SC) treatment to demonstrate a statistically significant diplopia (double vision) response in patients with chronic/inactive TED (24w diplopia responder rate 61% for Q4W elegro [p=0.0118] versus 55% for Q8W elegro [p=0.0419] versus 38% placebo).
Impact:
Potentially Best-in-class IGF-1R Inhibitor for TED: The data suggests that the Q8W (every 2 months) regimen is just as effective as the Q4W regimen, which would drastically reduce the treatment burden for patients. Nearly half (44%) of the patients in the Q4W arm achieved complete resolution of their diplopia (p = 0.0295), meaning they no longer experienced double vision in any direction of gaze. This is a potentially life-altering outcome for patients who previously relied on prisms or surgery.
Potentially Better Convenience: By offering an at-home autoinjector with “IV-like” efficacy, the trial sponsor aims to capture chronic patients who typically avoid the logistics of IV infusion centers.
Potentially Restores Confidence in Elegrobart’s Potency After Previous Mixed Phase 3: The successful Phase 3 REVEAL-2 data in chronic TED could revitalize enthusiasm elegrobart following the a mixed reaction to the Phase 3 REVEAL-1 active TED data on March 30, 2026. In the REVEAL-1 (active TED) readout, the proptosis responder rate (70.7% for the Q8W dose) was seen as slightly trailing the historical performance of IV Tepezza (~83% in its pivotal trial). This led to fears that the move from intravenous to subcutaneous (SC) delivery came with an “efficacy haircut.” REVEAL-2’s 54% responder rate in chronic TED is relatively strong, potentially allaying fears that the SC version is significantly weaker.
Next steps: The trial sponsor stated that they “remain on track to submit a Biologics License Application (BLA) to the U.S. FDA for elegrobart in Q1 2027”, with Phase 3 data for both inactive TED (REVEAL-2 trial) and active TED (REVEAL-1 trial) in hand. Their IV candidate, veligrotug (VRDN-001), has a PDUFA date of June 30, 2026. If approved, veligrotug’s launch could serve as the commercial foundation for a subsequent elegrobart launch, pending regulatory review.
To contact us, please send us an email at biotechreadout@gmail.com
Disclaimers
Investigational Status Disclaimer
The therapeutic candidates discussed in this newsletter are currently in clinical development and have not been approved for commercial sale by the U.S. Food and Drug Administration (FDA), the European Medicines Agency (EMA), or other global regulatory authorities. Their safety and efficacy have not been established. References to pipeline products and ongoing clinical trials involve significant risks and uncertainties. Statements regarding the potential safety, potency, or efficacy of investigational drugs reflect current hypotheses and are not a guarantee of future performance or regulatory clearance. The outcome of clinical trials is inherently unpredictable, and clinical results from earlier stages may not be predictive of results in later, larger-scale trials.
No Medical Advice Disclaimer
This newsletter is for informational and educational purposes only. The content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or another qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read in this publication.
No Patient-Provider Relationship Disclaimer
The information provided in this newsletter is for educational and analytical purposes only. Receipt of this information, or any interaction with this content, does not create a physician-patient, pharmacist-patient, or any other professional-provider relationship between you and the authors or publishers. This newsletter should not be used as a substitute for a personal consultation with a qualified healthcare professional.
Forward-Looking Statements Disclaimer
This newsletter contains “forward-looking statements” regarding future events, including clinical trial timing, regulatory milestones, and projected market performance. These statements are based on current expectations and assumptions that are subject to significant risks and uncertainties. Actual results may differ materially from those expressed or implied. We undertake no obligation to update these statements as a result of new information or future developments.
Third-Party Links & Content Disclaimer
This newsletter contains links to third-party websites, including clinical trial registries and corporate presentations. Biotech Readout does not endorse, guarantee, or assume responsibility for the accuracy or reliability of any information offered by third-party providers.
Errors and Omissions Disclaimer
While we strive for technical accuracy, the information in this newsletter is provided on an “as is” basis with no guarantees of completeness, accuracy, or timeliness. Biotech Readout assumes no liability for any errors or omissions in the content of this publication.
Non-Endorsement Disclaimer
Any reference to specific commercial products, processes, or services by trade name, trademark, or manufacturer does not constitute or imply an endorsement or recommendation by the author. All trademarks are the property of their respective owners.
No Investment Advice Disclaimer
This newsletter is for informational purposes only and does not constitute financial, investment, or legal advice. The author is not a registered investment advisor. You should consult with a professional financial advisor before making any investment decisions. The biotechnology sector is highly volatile; past performance is not indicative of future results.
Conflict of Interest Disclaimer
The author of this newsletter maintains a position of independence. At the time of publication, the author holds no direct financial interest, equity, or options in any of the companies mentioned in this report. No compensation has been received from any third party to feature or analyze specific therapeutic candidates or corporate entities.