
Eli Lilly to Acquire 4E Therapeutics
Ted Price's hunt for non-opioid painkillers pays off

Disclaimer: This newsletter is for educational and informational purposes only and does not constitute medical, investment, or financial advice, nor does it establish a provider-patient relationship. Content may include forward-looking statements and discussions of investigational therapeutic candidates that are not FDA/EMA approved; their safety and efficacy remain unestablished and clinical outcomes are unpredictable. While we strive for accuracy, all information is provided as is without guarantees. This newsletter is independent, and the author holds no financial positions in the companies mentioned nor receives third-party compensation for this coverage. Please find a complete version of our disclaimers at the bottom of this article and on our About page.

Introduction
On June 16, 2026, 4E Therapeutics, Inc. announced that it had been acquired by Eli Lilly and Company for an undisclosed amount. This acquisition centers on a preclinical pipeline of MNK inhibitors, developed as non-opioid painkillers:
4ET1103 (Neuropathic Pain): This is the company’s lead compound. It has completed a Phase 1 study, where it demonstrated a favorable safety profile.
4ET2124 (Migraine Pain): It is currently ready to begin IND-enabling studies.
MNK-eIF4E (Acute Pain): This compound is currently in the Discovery phase.
This acquisition signals Eli Lilly’s focus on complementary non-opioid peripheral pain mechanisms. On May 2025, Eli Lilly acquired SiteOne Therapeutics for up to $1 billion in cash, in-housing their Nav1.8 inhibitor STC-004, a voltage-gated sodium channel blocker designed to arrest electrical propagation in peripheral nerve. Lilly’s interest comes at an unprecedented time of need, as the United States grapples with the Opioid Crisis. From 1999-2023, approximately 806,000 people died in the United States from an opioid overdose according to the CDC, making it a national strategic priority.
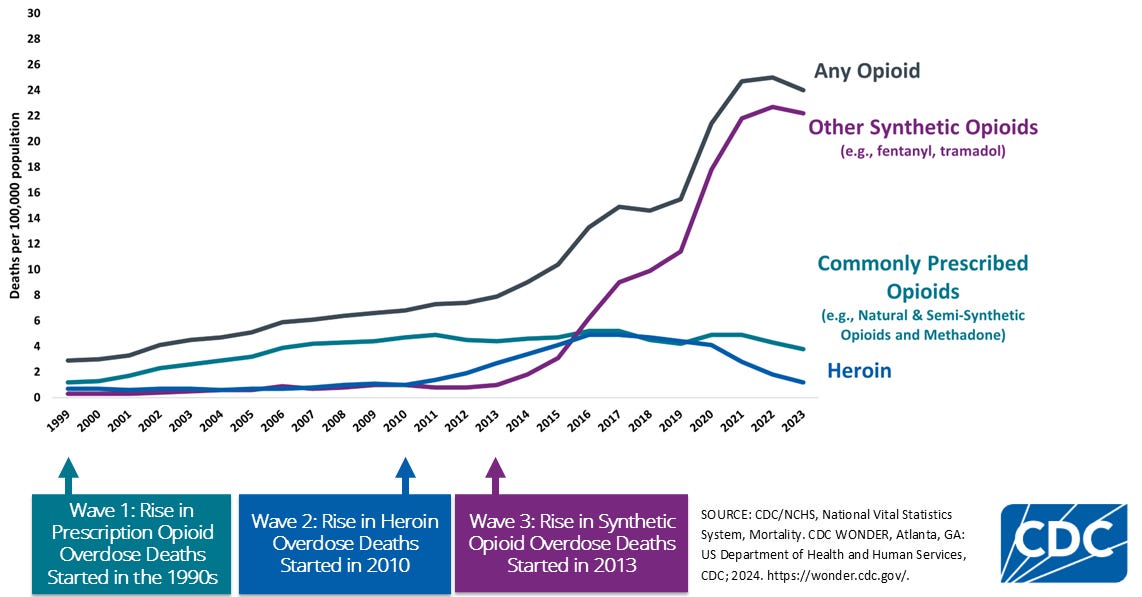
In this article, I will focus on how the pioneering work of Ted Price led to 4E Therapeutics and may someday help combat the Opioid Crisis.
We plan to discuss the Opioid Crisis and the rise of non-opioid painkillers in a future Frontiers in Medicine piece.
Lone Star Ranger
Growing up in Texas, Ted Price did not dream of slicing nerves or charting molecular pathways in a petri dish. Captivated by Carl Sagan’s Cosmos, he arrived at the University of Texas at Dallas (UTD) in the mid-1990s intending to become an astrophysicist. He wanted to map the stars, but science has a way of redirecting its most curious minds. Finding himself bogged down by the abstract mathematical architecture of advanced physics, Price sought counsel from his advisor. It was a serendipitous moment. UTD was quietly expanding a nascent neuroscience department, and his advisor nudged him to look inward rather than outward, suggesting that the human brain was an uncharted universe of its own. Price pivoted, becoming one of the very first undergraduates to graduate from UTD with a degree in neuroscience.
Afterwards, he moved on to the University of Texas Health Science Center at San Antonio for his Ph.D., immersing himself in pharmacology and the enigmatic behavior of peripheral pain, before packing his bags for a postdoctoral fellowship at McGill University in Montreal. It was at McGill, working alongside global leaders in pain mechanics, that Price began to challenge the orthodox medical view of chronic pain. For decades, medicine treated chronic pain as a wiring problem: an injury happens, a nerve fires, and the brain registers the signal. If the pain persisted, doctors assumed the injury hadn’t healed, or worse, that the patient was exaggerating. Price suspected the truth was far more insidious. He began investigating local translation machinery, the biological factories inside individual sensory neurons.
Through meticulous tracing, his lab discovered that a traumatic injury or a flood of inflammatory cytokines not only trigger an electrical impulse, but reprograms neurons through the MNK-eIF4E signaling pathway. In a healthy, uninjured state, baseline activity of the MNK-eIF4E pathway in sensory neurons is low. However, tissue injury, nerve damage, or metabolic distress triggers the pathway. Price’s work showed that the MNK pathway acts as a molecular crossroads where multiple, distinct upstream cascades converge in a three-step process:
The Cytokine/Growth Factor Influx: Peripheral injury floods the extracellular space with inflammatory cytokines (specifically IL-6 and TNF-alpha) and neurotrophins (like NGF).
The MAP Kinase Relays: These ligands bind to their respective receptors on nociceptors (such as gp130 for IL-6 and TrkA for NGF). This triggers two parallel intracellular signaling cascades: the ERK1/2 (extracellular signal-regulated kinase) pathway and the p38 MAPK pathway.
The MNK Convergence: Both active ERK and p38 physically bind to and phosphorylate Mitogen-Activated Protein Kinase-Interacting Kinases 1 and 2 (MNK1/2). Once phosphorylated, MNK1/2 becomes fully activated and primed to seek out its primary target: eIF4E.
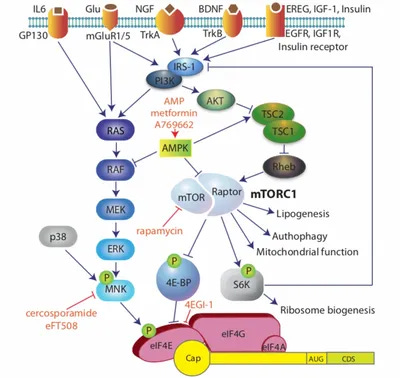
The crux of the Price Lab’s research centers on what happens once MNK1/2 is activated. MNK’s sole well-characterized role in this context is the highly specific phosphorylation of eIF4E (eukaryotic translation initiation factor 4E) at a single, precise amino acid residue: Serine 209 (Ser209). Before Price’s discoveries, it was generally assumed that phosphorylating eIF4E simply cranked up global protein synthesis across the board. The Price Lab showed that there was more nuance to the mechanism than initially appreciated. When MNK phosphorylates eIF4E, it selectively triggers the rapid synthesis of proteins that dictate neuronal plasticity and pain hypersensitivity. These changes convert a normal, protective sensory alert system into a permanently damaged, hyper-reactive chronic circuit in three ways:
Ion Channel Sinking: The newly translated proteins facilitate the rapid trafficking and insertion of voltage-gated sodium channels (like Nav1.7 and Nav1.8) and transient receptor potential channels (like TRPV1) directly into the nociceptor’s plasma membrane.
Threshold Drop: With a vastly higher density of these channels embedded in the membrane, the resting membrane potential changes. The physical threshold required to trigger an action potential plummets.
Allodynia and Hyperalgesia: Light touches that should feel benign (like clothing resting against the skin) are suddenly amplified into intense pain signals (allodynia). Minor painful stimuli are registered as excruciating (hyperalgesia).
While Price initiated his earliest exploratory research into localized protein synthesis while he was a postdoc at McGill (from 2004 to 2007), the heavy-lifting discovery, mapping, and human validation of the MNK-eIF4E axis occurred entirely after he returned to Texas and established his own independent labs. In 2014, Price was recruited back to his alma mater, UTD, as a star faculty member. Five years later in 2019, he became the founding director of the Center for Advanced Pain Studies (CAPS).
Of Mice and Men
In July 2017, Price published a foundational paper in The Journal of Neuroscience entitled “The MNK-eIF4E Signaling Axis Contributes to Injury-Induced Nociceptive Plasticity and the Development of Chronic Pain”. Prior to this publication, it was well known that activity-dependent mRNA translation via the mTORC1 and MAPK pathways drove pain sensitization. However, these upstream pathways are massive, complex, and regulate thousands of cellular functions. The Price Lab set out to isolate the exact step where these pathways converge to dictate hyperalgesic priming, or “pain memory” caused by molecular changes in neurons increase pain sensitivity. They hypothesized that the precise phosphorylation of the 5’ cap-binding protein eIF4E by its specific upstream kinase, MNK1/2, was the exact molecular switch responsible.
To investigate their hypothesis, the researchers designed a series of parallel experiments testing both genetic and pharmacological interventions across both male and female mice, confirming that the pathway operates identically across sexes. These studies involved the following key perturbations:
eIF4ES209A Knock-In Mouse: This was the study’s crown jewel. The team utilized mice engineered with a single point mutation that converted Serine 209 to Alanine (S209A). These mice have perfectly functional eIF4E proteins, but their eIF4E cannot be phosphorylated by MNK.
Mnk1/2-/- Knockout Mouse: These were mice completely lacking the genes that encode the MNK1 and MNK2 kinases.
Cercosporamide: A small-molecule inhibitor of MNK1/2 used as a tool compound to see if the genetic models could be replicated chemically in wild-type (WT) animals.
Their first finding involved the injection of potent pain-stimulating and inflammatory agents, specifically Nerve Growth Factor (NGF), Interleukin-6 (IL-6), and carrageenan, into the hindpaws of the mice. In wild-type mice, the injections caused acute mechanical and thermal hypersensitivity, which eventually subsided. A subsequent injection of a low-dose inflammatory mediator (like PGE2) caused an immediate, severe, and prolonged return of pain behaviors. This fits the definition of hyperalgesic priming, cell signaling changes that sensitize mice to chronic pain. eIF4ES209A Knock-In and Mnk1/2-/- Knockout mice displayed normal acute reflexes to the initial injury, proving their baseline sensory warning systems were completely intact. However, they showed a profound reduction in secondary mechanical and thermal hypersensitivity, decreased affective pain behaviors (measured via facial grimacing scales), and completely failed to trigger molecular signaling associated with hyperalgesic priming. The biological memory of the pain couldn’t be formed so long as the interaction between eIF4 and Mnk1/2 was absent. The paper also looked at peripheral nerve injury (PNI) via a surgical model. They discovered that the intense, long-lasting cold hypersensitivity typically brought on by direct structural nerve damage was heavily attenuated in both the eIF4ES209A Knock-In and Mnk1/2-/- Knockout mice, as well as in WT mice treated systemically with cercosporamide.
To understand why the behavior changed, the team performed whole-cell patch-clamp recordings and Ca2+ imaging on small-diameter dorsal root ganglion (DRG) neurons, the primary pain-sensing cells. Exposing wild-type DRG neurons to NGF or IL-6 caused a massive surge in ramp current-evoked action potential spiking. The nerves became hyper-excitable, firing rapidly at minor stimulation thresholds. Neurons isolated from eIF4ES209A mice showed almost no increase in excitability when exposed to NGF or IL-6. The threshold for firing remained at healthy, baseline levels. Similarly, treatment of wild-type DRG neurons with the MNK inhibitor cercosporamide blocked the hyperexcitability induced by NGF and IL-6, matching the genetic mouse findings.
A subsequent 2023 bioRxiv preprint entitled “MNK inhibitor eFT508 (Tomivosertib) suppresses ectopic activity in human dorsal root ganglion neurons from dermatomes with radicular neuropathic pain” bridged the mouse-to-human translational gap. A key part of this study lies in its procurement and utilization of viable human tissue, a logistical feat managed by the Center for Advanced Pain Studies (CAPS) at UT Dallas in collaboration with surgical teams, such as those at MD Anderson Cancer Center. The team recovered human dorsal root ganglion (DRG) tissue from patients undergoing complex thoracic vertebrectomy surgeries (procedures involving the partial removal of vertebrae to relieve severe spinal cord or dorsal root compression). The tissue was harvested specifically from patients with radiculopathy (pinched, inflamed spinal nerves causing severe, radiating nerve pain along designated dermatomes). These neurons were compared directly against healthy DRG tissue recovered from organ donors who did not have a history of chronic neuropathic pain.
Once the human DRGs were surgically extracted, the Price Lab immediately transported and dissociated the tissue to culture live human sensory neurons. They utilized whole-cell patch-clamp electrophysiology to record the real-time electrical signatures of these cells. In preclinical models, a damaged nerve cell generates Spontaneous Activity (SA) or ectopic activity, meaning it fires action potentials continuously without any external heat, touch, or chemical trigger. This paper sought to determine whether human radiculopathy neurons possessed this same pathological firing and if an MNK inhibitor could silence it. When researchers applied 25 nM of Tomivosertib (a MNK1/2 inhibitor) directly onto the firing human radiculopathy neurons, it suppressed the pathological spontaneous firing within minutes. Crucially, when the drug was washed out of the cellular bath, the spontaneous activity slowly returned over time. This demonstrated a direct, reversible pharmacological engagement rather than a toxic, permanent killing of the cell.
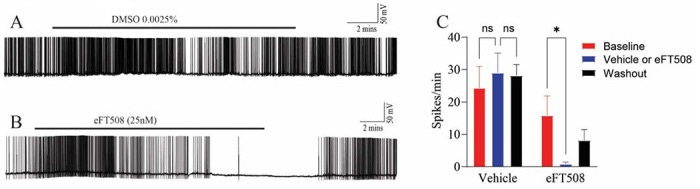
By studying the precise shape of the action potentials before and after tomivosertib application, the team characterized how acute translation blockade alters electrical propagation:
Decreased Action Potential Amplitude: The peak height of the nerve’s electrical spike shrank.
Altered Afterhyperpolarization (AHP) Currents: The magnitude of the resetting current after a cell fires was significantly modified.
The Ion Channel Connection: These changes strongly indicated that blocking MNK immediately modifies the functional activity and deployment of voltage-gated Sodium (Na+) and Potassium (K+) channels embedded in the human pain nerves.
Spurring it Forward
Ted Price never believed that a scientist’s job ended with a published paper. He openly maintained that if you discover a way to alleviate human suffering, you have a moral obligation to get it to patients. This spirit, combined with high-quality neurobiology, led him to serial entrepreneurship.
First came CerSci Therapeutics, a company he co-founded to develop small-molecule catalysts for neuropathic pain. CerSci was founded in 2015 alongside his frequent UT Dallas collaborator Dr. Gregory Dussor and then-graduate student Lucas Rodriguez, who drafted the business plan during an entrepreneurship course. The company was built to intercept acute and postoperative pain before it could necessitate opioid exposure. CerSci acquired and optimized a lead small-molecule compound, CT-044, a novel peroxynitrite decomposition catalyst. Rather than dulling pain centrally in the brain or binding to dangerous opioid receptors, the molecule worked peripherally to damp down the oxidative stress and inflammatory cascades that cause nociceptors to flare post-surgery. After raising nearly $20 million in venture capital and securing essential NIH Small Business Innovation Research (SBIR) grants, CerSci proved the safety and mechanics of its platform. In August 2020, the startup was acquired by Acadia Pharmaceuticals for $52.5 million upfront, with total development and commercial milestones pushing the deal’s potential value up to $525 million.
Following the acquisition of CerSci, Price co-founded 4E Therapeutics to commercialize his lab’s definitive breakthrough: the MNK-eIF4E translational axis. Led by CEO T. Craig Benson and VP of Drug Development James Sahn, the startup aimed to silence the biological printer that writes chronic pain memory into sensory nerves. While systemic MNK inhibitors had been tested in oncology, they crossed the blood-brain barrier, triggering central toxicities like depression. 4E Therapeutics engineered 4ET1103, a highly optimized, peripherally restricted oral small molecule MNK inhibitor. By isolating the drug’s effects strictly to the dorsal root ganglia (DRG), they could arrest pathological protein translation while minimizing central nervous system or addictive side effects. After advancing 4ET1103 through a Phase 1 trial, Eli Lilly agreed to acquire 4E Therapeutics and absorb its pipeline of MNK inhibitors into their expanding non-opioid pain portfolio.
Recognizing that the standard industry practice of using rodent models was the primary reason most pain drugs failed in clinical trials, Price co-founded Doloromics. Doloromics functions as a proprietary, data-driven drug discovery platform. Instead of testing random compounds on mice, the company leverages the massive, high-resolution human datasets generated by the Center for Advanced Pain Studies (CAPS). By utilizing comparative human transcriptomics, specifically sequencing healthy human DRGs against tissue from donors who suffered from chronic conditions like diabetic neuropathy, Doloromics employs machine learning to identify entirely new, highly specific cell-surface targets unique to human pain pathways, licensing these pristine targets out to broader pharmaceutical developers.
Demonstrating his agility across different commercial scales, Price also launched Ted’s Brain Science Products to address localized pain at the skin level. Born out of his early academic research into the metabolic management of pain, this venture commercializes over-the-counter topical pain creams. The formulations utilize resveratrol and metformin to activate AMP-activated protein kinase (AMPK). Price’s lab had previously discovered that AMPK acts as a natural brake on nociceptor amplification. By delivering these activators topically, the cream helps reset hyperactive nerve endings in the skin without systemic drug interaction.
Conclusion
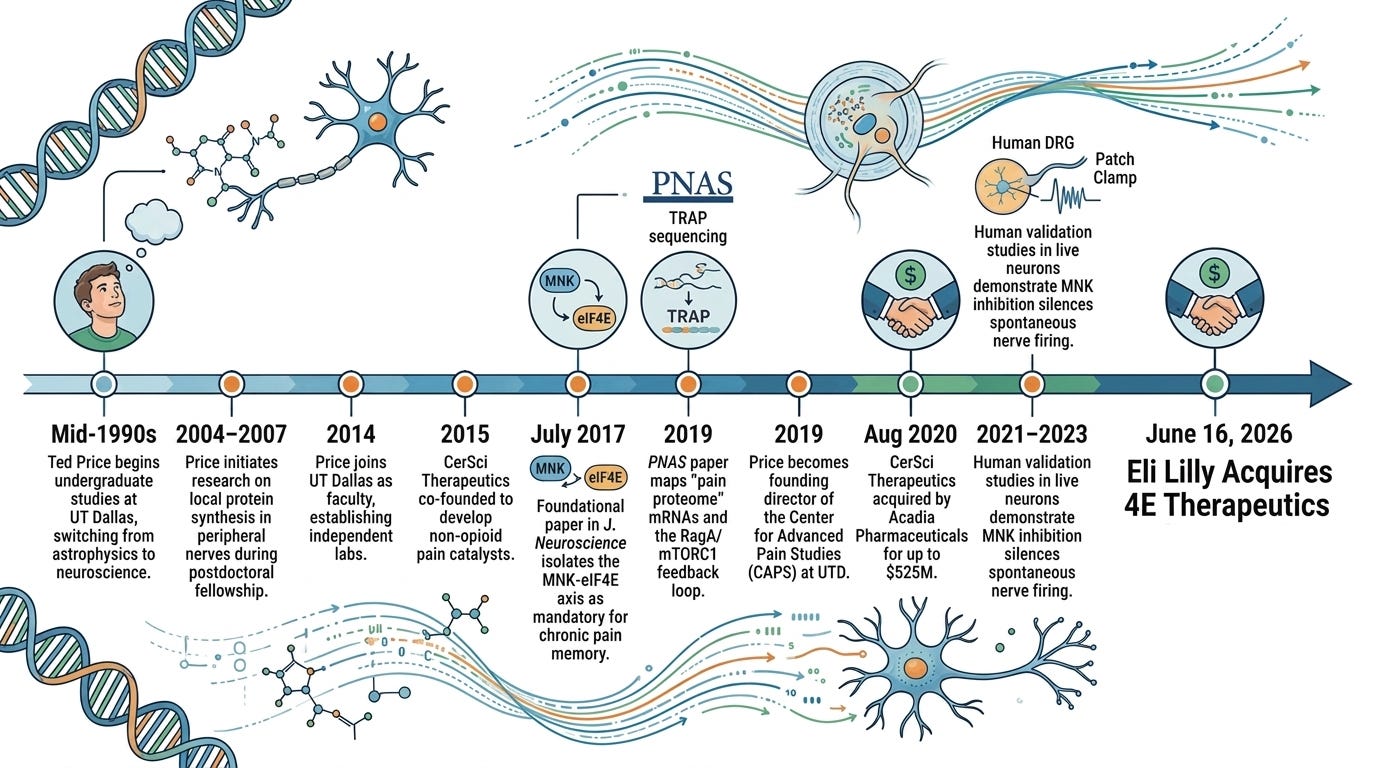
Eli Lilly’s acquisition of 4E Therapeutics is a landmark in a thirty-year scientific journey that began when an undergraduate student in Dallas pivoted from astronomy to neuroscience. By backing Dr. Ted Price’s vision, Lilly is betting on a new mechanism for chronic pain. For decades, the pharmaceutical industry tried to solve pain by overriding the brain with central opioids, an approach that triggered a devastating public health crisis. As 4E Therapeutics enters into a collaboration with a global pharmaceutical giant, the industry moves one step closer to a future where chronic pain is no longer a permanent sentence, but a neuronal memory that can be rewritten.
To contact us, please send us an email at biotechreadout@gmail.com
Disclaimers
Investigational Status Disclaimer
The therapeutic candidates discussed in this newsletter are currently in clinical development and have not been approved for commercial sale by the U.S. Food and Drug Administration (FDA), the European Medicines Agency (EMA), or other global regulatory authorities. Their safety and efficacy have not been established. References to pipeline products and ongoing clinical trials involve significant risks and uncertainties. Statements regarding the potential safety, potency, or efficacy of investigational drugs reflect current hypotheses and are not a guarantee of future performance or regulatory clearance. The outcome of clinical trials is inherently unpredictable, and clinical results from earlier stages may not be predictive of results in later, larger-scale trials.
No Medical Advice Disclaimer
This newsletter is for informational and educational purposes only. The content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or another qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read in this publication.
No Patient-Provider Relationship Disclaimer
The information provided in this newsletter is for educational and analytical purposes only. Receipt of this information, or any interaction with this content, does not create a physician-patient, pharmacist-patient, or any other professional-provider relationship between you and the authors or publishers. This newsletter should not be used as a substitute for a personal consultation with a qualified healthcare professional.
Forward-Looking Statements Disclaimer
This newsletter contains “forward-looking statements” regarding future events, including clinical trial timing, regulatory milestones, and projected market performance. These statements are based on current expectations and assumptions that are subject to significant risks and uncertainties. Actual results may differ materially from those expressed or implied. We undertake no obligation to update these statements as a result of new information or future developments.
Third-Party Links & Content Disclaimer
This newsletter contains links to third-party websites, including clinical trial registries and corporate presentations. Biotech Readout does not endorse, guarantee, or assume responsibility for the accuracy or reliability of any information offered by third-party providers.
Errors and Omissions Disclaimer
While we strive for technical accuracy, the information in this newsletter is provided on an “as is” basis with no guarantees of completeness, accuracy, or timeliness. Biotech Readout assumes no liability for any errors or omissions in the content of this publication.
Non-Endorsement Disclaimer
Any reference to specific commercial products, processes, or services by trade name, trademark, or manufacturer does not constitute or imply an endorsement or recommendation by the author. All trademarks are the property of their respective owners.
No Investment Advice Disclaimer
This newsletter is for informational purposes only and does not constitute financial, investment, or legal advice. The author is not a registered investment advisor. You should consult with a professional financial advisor before making any investment decisions. The biotechnology sector is highly volatile; past performance is not indicative of future results.
Conflict of Interest Disclaimer
The author of this newsletter maintains a position of independence. At the time of publication, the author holds no direct financial interest, equity, or options in any of the companies mentioned in this report. No compensation has been received from any third party to feature or analyze specific therapeutic candidates or corporate entities.