
China achieves world's first CAR-T approval for solid tumors, green shoots in neuropsych, and more
Weekly Readout #11: Week ending June 26, 2026

Disclaimer: This newsletter is for educational and informational purposes only and does not constitute medical, investment, or financial advice, nor does it establish a provider-patient relationship. Content may include forward-looking statements and discussions of investigational therapeutic candidates that are not FDA/EMA approved; their safety and efficacy remain unestablished and clinical outcomes are unpredictable. While we strive for accuracy, all information is provided as is without guarantees. As of the date of publication, the author holds no direct equity positions in the specific companies mentioned in this issue nor receives third-party compensation for this coverage. Please find a complete version of our disclaimers at the bottom of this article and on our About page.

Press ⬇️ to go down to a section and ⬆️ to go back up to the Table of Contents
Table of Contents
This week, we discuss:
China Biotech
Acquisitions
AbbVie to Acquire Apogee, Paragon’s long-acting atopic dermatitis & asthma pipeline pays off ⬇️
Approvals
Clinical Trial Data
China Biotech
World’s first CAR-T approval for solid tumors
In a historic development for CAR-T, the National Medical Products Administration (NMPA) has approved CARsgen’s satri-cel (satricabtagene autoleucel), a CAR-T cell therapy targeting Claudin 18.2, for use in China. It is indicated for patients with Claudin 18.2-positive, HER2-negative gastric cancer or gastroesophageal junction adenocarcinoma (GC/GEJC). For me, the NMPA approval of satri-cel marks the precise moment when China Biotech officially became innovators, rather than imitators. Sure, Summit/Akeso’s PD1 x VEGF bispecific antibody ivonescimab has generated a lot of buzz, but its OS advantage over Keytruda hasn’t yet been established in U.S. patients. The approval of satri-cel in China is a first on two fronts:
First Solid Tumor CAR-T Approval Worldwide: This marks the first time any regulatory body in the world has approved a CAR-T cell therapy for a solid tumor indication. Historically, approved cell therapies have been restricted to hematologic malignancies (such as leukemia, lymphoma, and multiple myeloma). To break through the solid tumor microenvironment, CARsgen utilized a unique, optimized preconditioning regimen prior to satri-cel infusion. This added low-dose nab-paclitaxel to the standard cyclophosphamide and fludarabine (Cy/Flu) lymphodepletion backbone, which was designed to enhance CAR-T cell infiltration and anti-tumor durability.
First Approved Claudin 18.2 Targeting Therapy: This approval represents the global debut of a therapeutic specifically targeting Claudin 18.2 (CLDN18.2), a highly validated and sought-after biomarker in gastrointestinal cancers.
Led by Professor Lin Shen at Peking University Cancer Hospital, the study evaluated 156 patients with heavily pretreated, Claudin 18.2+ HER2- advanced gastric or gastroesophageal junction adenocarcinoma (GC/GEJC) who had progressed on at least two prior lines of therapy. Patients were randomized 2:1 to receive either satri-cel (up to 3 infusions of 250 × 106 cells) or treatment of physician’s choice (TPC), which included standard chemotherapies or an anti-PD-1 antibody. The trial demonstrated a statistically significant and clinically meaningful benefit across both primary and secondary endpoints. Satri-cel nearly doubled the median PFS compared to standard care, achieving 3.25 months versus 1.77 months in the control arm (HR = 0.37, p < 0.0001). Median overall survival was significantly extended to 7.92 months with satri-cel, compared to 5.49 months for patients receiving standard therapies (HR = 0.693, p = 0.0416). True to the nature of CAR-T cell therapies, CRS occurred in 95% of treated patients, though it was characterized as “manageable”. Grade 3 or higher treatment-emergent adverse events (TEAEs) occurred in 99% of the satri-cel arm (vs. 63% in the TPC arm), primarily driven by predictable lymphopenia (98%), leukopenia (77%), and neutropenia (66%) resulting from the lymphodepletion regimen.
Sources: CARsgen press release, OncLive article
BIO board met with U.S. national security & health officials to discuss China
Endpoints broke the news that the board of the Biotechnology Innovation Organization (BIO), led by CEO John Crowley, met directly with US national security and health officials to discuss the biotech industry’s evolving relationship with China. For context, Biotechnology Innovation Organization (BIO) is the world’s largest trade association representing more than 1,000 members in the biotechnology industry. It is based in Washington, D.C. and it serves as a powerful advocacy group, political lobby, and networking hub for the life sciences sector. Under Crowley’s leadership, the major biotech trade group has taken a decidedly patriotic turn, leaning heavily into national security. Crowley has previously characterized the dependence on Chinese contract manufacturers as a “Sputnik moment” for the US to regain control over its medical supply chain and manufacturing capabilities. The meeting aligns with BIO’s dramatic organizational about-face to support the controversial Biosecure Act. This legislative push aims to restrict US biotech companies from working with specific Chinese contract research and manufacturing entities (like WuXi AppTec). While Crowley has emphasized that the goal is not a wholesale “decoupling” from China, the strategy is explicitly designed to address congressional anxieties regarding intellectual property, data security, and geopolitical reliance.

Acquisitions

Approvals
Ionis - Tryngolza (APOC3 ASO) / Approval (sHTG)
The FDA has granted a major label expansion for Tryngolza (olezarsen), an antisense oligonucleotide (ASO) targeting APOC3. Originally approved for the ultra-rare familial chylomicronemia syndrome (FCS), Tryngolza is now approved as an adjunct to diet to reduce triglycerides and the risk of acute pancreatitis in adults with Severe Hypertriglyceridemia (sHTG; defined as triglycerides ≥500 mg/dL) and will be available for sHTG in the U.S. starting in July. The expansion is supported by robust data from two pivotal Phase 3 trials (CORE and CORE2), which enrolled over 1,000 patients. The trials demonstrated dramatic, placebo-adjusted triglyceride reductions (up to 72% at the 80 mg monthly dose) and a critical 85% reduction in the risk of acute pancreatitis episodes. This shifts Tryngolza from a niche rare-disease drug (FCS has ~3,000 U.S. patients according to Ionis) to a broad commercial therapeutic with more than a million patients in the U.S. with “high risk” (defined by Ionis as “triglycerides ≥800 mg/dL or triglycerides ≥500 mg/dL + acute pancreatitis and/or comorbidities”). Close competition is on the horizon. Arrowhead Pharmaceuticals (ARWR) is expected to file its competing APOC3-targeted RNAi therapeutic, Redemplo, for sHTG by the end of 2026.
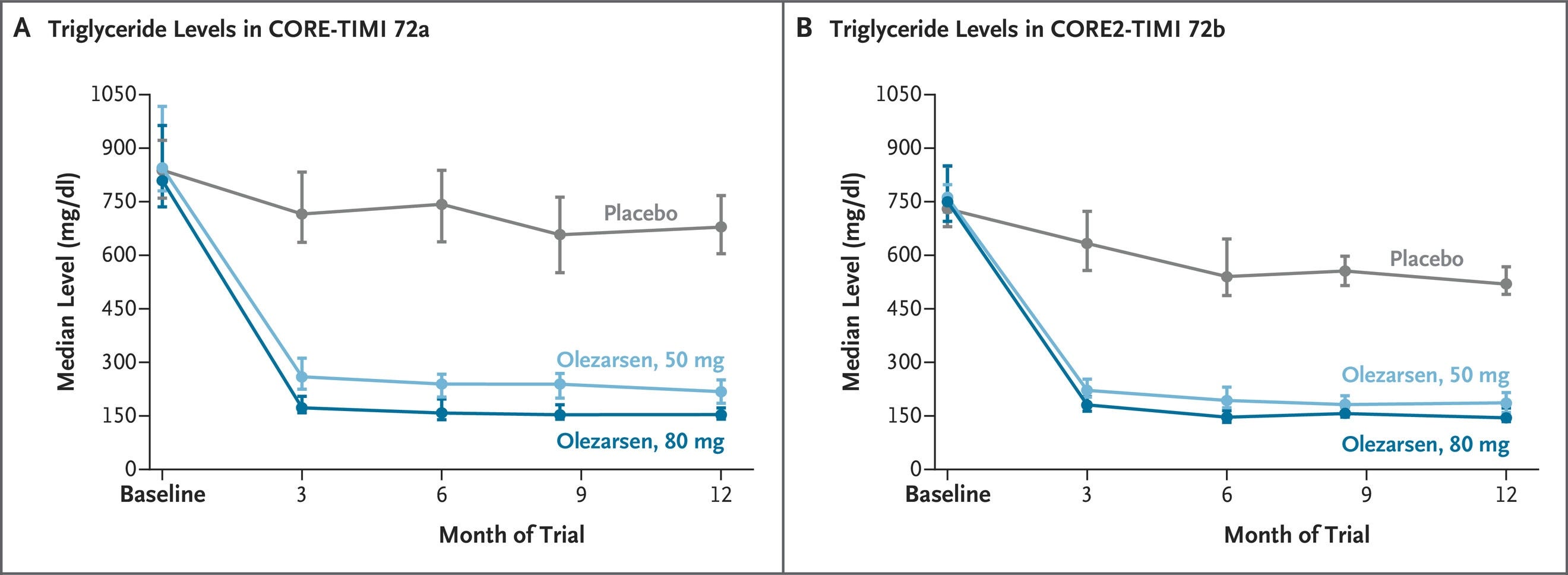
Sources: Ionis press release, Ionis presentation, new Tryngolza label, NEJM paper
Gilead - Trodelvy (TROP2 ADC) / Approval (1L TNBC)
The FDA has granted a landmark dual label expansion for Trodelvy, a first-in-class TROP2-directed antibody-drug conjugate (ADC). Moving up from its established late-line indications, Trodelvy is now approved for the first-line (1L) treatment of adults with unresectable locally advanced or metastatic triple-negative breast cancer (mTNBC) across all patient populations regardless of PD-L1 status. The broad frontline label explicitly covers two distinct metastatic patient populations, each supported by a Phase 3 clinical trial:
Monotherapy for patients who are not candidates for checkpoint inhibitor (PD-1/L1) therapy. This was supported by the Phase 3 ASCENT-03 trial, which enrolled 558 checkpoint-ineligible patients. Trodelvy monotherapy demonstrated a median progression-free survival (mPFS) of 9.7 months vs. 6.9 months for physician’s choice chemotherapy, reflecting a 38% reduction in the risk of disease progression or death (HR = 0.62; p < 0.0001).
Combination with Merck’s Keytruda (pembrolizumab, including the subcutaneous Keytruda Qlex) for patients with PD-L1-positive tumors (CPS ≥10). This was supported by the Phase 3 ASCENT-04 / KEYNOTE-D19 trial, which enrolled 443 PD-L1+ patients. Trodelvy + Keytruda extended mPFS to 11.2 months vs. 7.8 months for chemo + Keytruda, reflecting a 35% risk reduction (HR = 0.65; p = 0.0009). Overall survival (OS) data for both trials remained immature at the time of analysis.
Since over half of mTNBC patients historically never survive or progress to receive a second line of therapy, establishing an ADC-based regimen in the frontline setting represents a significant paradigm shift in the standard of care. The National Comprehensive Cancer Network (NCCN) immediately incorporated both regimens as Category 1 preferred frontline options. Trodelvy‘s approval follows closely behind AstraZeneca and Daiichi Sankyo’s TROP2 ADC Datroway (datopotamab deruxtecan), which was approved for the checkpoint-ineligible frontline setting. However, only Trodelvy’s label has the broader inclusion of the PD-L1+ combo population.
Source: Gilead press release, new Trodelvy label
Clinical Trial Data
Absci - ABS-201 (anti-PRLR mAb) / Phase 1 (androgenetic alopecia)
Absci has announced highly encouraging interim Phase 1 data from its HEADLINE trial evaluating ABS-201, an AI-designed, long-acting monoclonal antibody targeting the prolactin receptor (PRLR).
Androgenetic alopecia (AGA) is primarily driven by genetic hypersensitivity to dihydrotestosterone (DHT), which causes follicular miniaturization, a shortened anagen growth phase, and progressive hair thinning. While the standard of care relies on daily treatments like oral finasteride to suppress DHT and topical minoxidil to boost follicular blood flow, Absci’s ABS-201 introduces a novel mechanism as an AI-designed, long-acting monoclonal antibody targeting the prolactin receptor (PRLR). By blocking localized prolactin signaling, which otherwise triggers hair follicle stem cell apoptosis and forces follicles into dormancy, ABS-201 is designed to the progenitor cell pool and prolongs the active anagen phase to restore terminal hair growth.
The ongoing Phase 1/2a HEADLINE trial evaluated Absci’s antibody, ABS-201, in a randomized, placebo-controlled single ascending dose (SAD) study of 32 healthy volunteers across four intravenous cohorts (150 mg to 1800 mg). Data from the June 8, 2026 cutoff demonstrated that the biologic was well-tolerated with zero serious adverse events and mostly mild treatment-related effects (primarily headaches). Crucially, ABS-201 exhibited an exceptionally prolonged half-life of at least 65 days with no anti-drug antibody impact, supporting a highly convenient maintenance dosing schedule of just one injection every two to three months. This profile validates Absci’s de novo generative AI platform by proving a fully AI-designed antibody can achieve optimal, non-immunogenic drug properties in humans, while offering a potential long-acting therapeutic alternative that, if approved, could alleviate the daily compliance burden and side-effect profiles of current standard-of-care treatments like minoxidil and finasteride
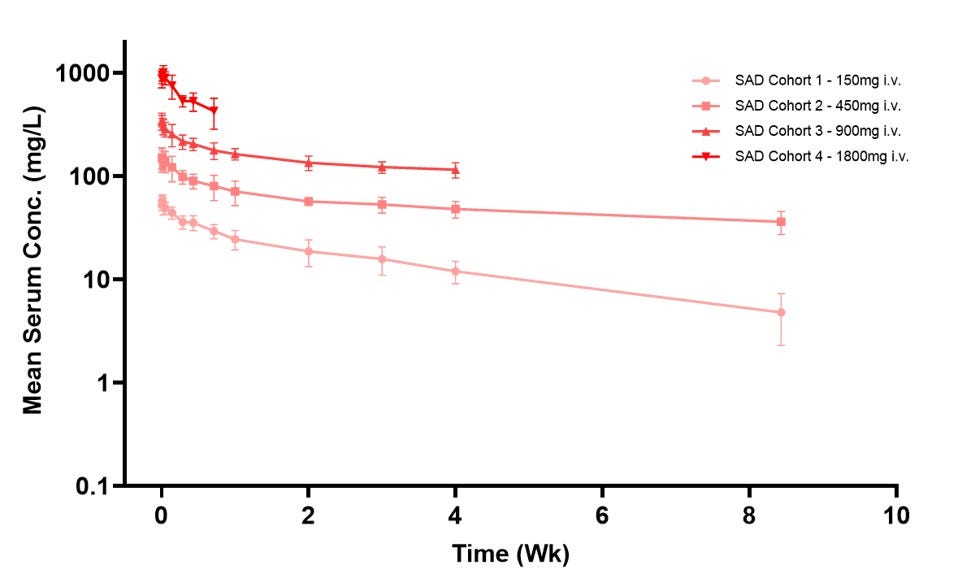
Following Safety Review Committee approval, the HEADLINE trial has immediately advanced into the Subcutaneous (SC) Multiple Ascending Dose (MAD) phase in patients with active AGA. Dosing cohorts are set at 300 mg, 600 mg, and 1200 mg SC to establish real-world, patient-friendly administration parameters. Absci plans to report interim clinical proof-of-concept (PoC) data from the SC MAD cohorts in the second half of 2026 and full clinical proof-of-concept data in early 2027. Since prolactin signaling is a known driver of endometrial lesions and pelvic pain sensitivity, Absci plans to advance ABS-201 into a global Phase 2 trial for endometriosis by the end of 2026. Pending FDA interactions on the MAD dose-ranging data, Absci aims to leapfrog directly into two global Phase 3 studies in male-pattern hair loss by late 2027.
Sources: Absci press release
Definium - DT120 (LSD ODT) / Phase 3 (MDD)
Definium Therapeutics has announced successful topline results from its pivotal Phase 3 Emerge trial evaluating DT120 ODT, an orally disintegrating tablet formulation of lysergide tartrate, for the treatment of Major Depressive Disorder (MDD).
Major Depressive Disorder (MDD) is a complex neuropsychiatric condition characterized by persistent depressed mood and anhedonia, pathophysiologically driven by neural circuitry dysfunction, monoamine depletion, and impaired neuroplasticity. While the current standard of care relies on chronic daily pharmacotherapy, such as SSRIs or SNRIs, and psychotherapy to gradually alter neurotransmitter levels, Definium’s DT120 ODT (lysergide tartrate) introduces a novel approach as an optimized, orally disintegrating tablet (ODT) formulation of LSD. Functioning as a potent 5-HT2A (serotonin 2A) receptor agonist, a single acute administration of DT120 triggers rapid downstream intracellular signaling pathways that drive an immediate burst of cortical synaptogenesis and structural neural plasticity, with the goal of resetting disrupted frontostriatal networks to restore mood and cognitive flexibility without requiring daily dosing.
The pivotal Phase 3 Emerge trial (NCT06941844) evaluated a single 100 µg dose of DT120 ODT against placebo in 149 adults with moderate-to-severe Major Depressive Disorder (MDD), in an effort to isolate the drug’s organic pharmaceutical effect without adjunctive psychotherapy. The study met its primary endpoint at Week 6, achieving a statistically significant (p<0.0001) and clinically meaningful placebo-adjusted LS mean reduction in MADRS total score of -8.1 points (−13.3 vs. −5.2 for placebo), alongside a 35% response rate and 24% remission rate. Secondary endpoints highlighted a rapid onset at Week 1 (−14.2 point placebo-adjusted reduction) and sustained durability out to Week 12 (−7.3 point placebo-adjusted reduction). DT120 ODT was generally well tolerated with no new safety signals or increased suicidal ideation. Approximately 99% of adverse events were mild-to-moderate, transient, and limited to the dosing day, with all patients successfully clearing clinic monitoring within 8 hours (mean: 5.8 hours). By delivering rapid, quarter-long depression relief from a single acute session, DT120 ODT could shift the treatment paradigm away from burdensome daily oral therapies and differentiates itself from existing acute options like J&J’s Spravato, which requires ongoing bi-weekly maintenance visits and averages a numerically smaller cross-trial MADRS delta ( -6.8 points versus -8.1 points for DT120).
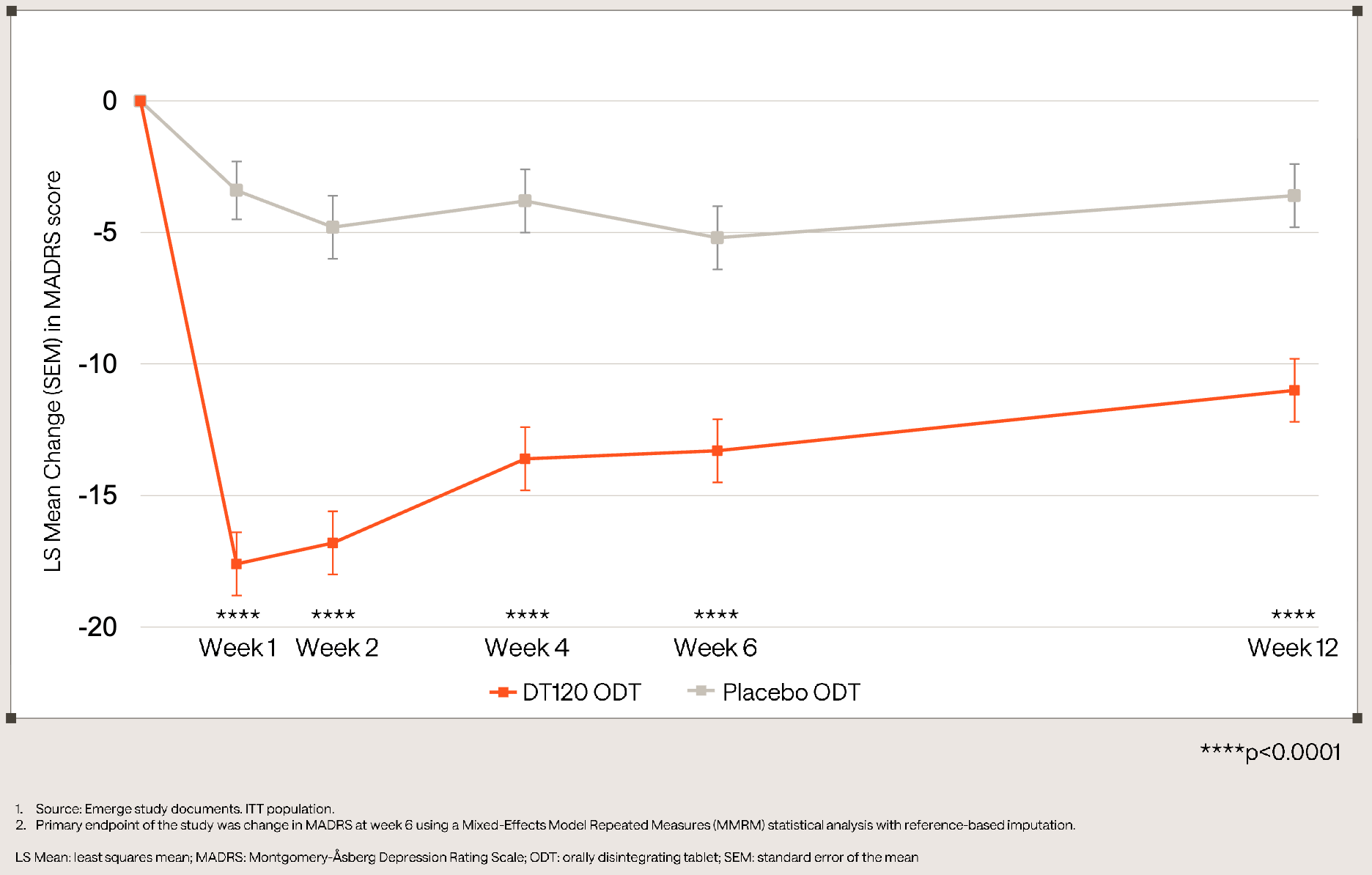
Definium is currently running Ascend (NCT07592689), a second late-stage Phase 3 trial in MDD exploring safety and varying doses in ~175 patients to complete its registration package for the FDA. Topline Phase 3 data in Generalized Anxiety Disorder (GAD) from the Panorama and Voyage studies are anticipated in Q3 2026. Definium plans to initiate its Phase 3 Haven study evaluating DT120 in Post-Traumatic Stress Disorder (PTSD) in early 2027.
Sources: Definium press release, Definium slide deck
Otsuka - Centanafadine (SNDRI) / Phase 3b (ADHD)
Otsuka presented highly statistically significant Phase 3b data for centanafadine, demonstrating a reduction in ADHD symptoms compared to placebo in adults with comorbid anxiety ahead of its July 24, 2026 PDUFA date.
Attention-Deficit/Hyperactivity Disorder (ADHD) is a neurodevelopmental condition driven by a dysregulation of dopamine and norepinephrine signaling in the prefrontal cortex, which impairs key executive functions like impulse control and sustained attention. While the standard of care primarily relies on central nervous system stimulants (such as methylphenidate or amphetamines) to boost these neurotransmitters, or non-stimulants like atomoxetine for patients who cannot tolerate them, Otsuka’s centanafadine introduces a novel approach as a first-in-class Norepinephrine, Dopamine, and Serotonin Reuptake Inhibitor (NDSRI). By simultaneously inhibiting the reuptake of all three major monoamine neurotransmitters, this triple reuptake inhibitor aims to deliver robust clinical efficacy with a favorable tolerability profile and a lower potential for abuse than traditional stimulants.
The Phase 3b study (NCT06973577) evaluated once-daily centanafadine XR (280 mg) over 8 weeks in 315 adults with ADHD and comorbid anxiety, a historically underserved population that comprises up to 50% of adult ADHD cases. The trial met its primary endpoint, demonstrating a statistically significant early-onset reduction in ADHD symptoms by Week 8 (LS mean change in AISRS of -18.5 versus -12.6 for placebo; p < 0.0001), alongside significant improvements in anxiety symptoms (HAM-A placebo-adjusted reduction of 1.92 points). The treatment was generally well tolerated with a safety profile consistent with previous trials, characterized by a low potential for abuse and common adverse events like decreased appetite and headache. Ultimately, these data demonstrate encouraging preliminary evidence of simultaneous efficacy on both core ADHD and anxiety symptoms, yielding a -5.87 placebo-adjusted AISRS treatment difference that numerically surpasses Otsuka’s earlier 6-week Phase 3 trials and validates the extended 8-week extended-release strategy, though direct cross-trial comparisons carry inherent limitations due to differing study designs and patient populations.
The FDA is currently reviewing centanafadine’s New Drug Application (NDA) under Priority Review across pediatric, adolescent, and adult ADHD populations. The PDUFA target action date is July 24, 2026. Otsuka is actively building out its commercial infrastructure and training sales forces ahead of the looming decision. While the drug has shown low abuse liability in trials, a standard Drug Enforcement Administration (DEA) scheduling review is expected post-approval, which could delay the actual commercial launch by approximately three months, according to Otsuka.
Sources: Otsuka press release
Verastem - VS-7375 (KRAS G12D inhibitor) / Phase 1/2 (KRAS G12D+ solid tumors)
Verastem Oncology has announced preliminary anti-tumor activity from its Phase 1/2 mixed clinical trial evaluating VS-7375, an oral, highly selective small-molecule KRAS G12D inhibitor, in patients with advanced solid tumors.
The KRAS G12D mutation is a prevalent oncogenic driver occurring in 30-45% of pancreatic ductal adenocarcinoma (PDAC) cases, where a missense mutation locks the KRAS protein in a constitutively active state that hyperactivates downstream MAPK and PI3K-AKT pathways to drive uncontrolled cell proliferation, metabolic reprogramming, and survival. Since the structural complexity of the G12D pocket has historically limited targeted therapy options, the standard of care for advanced solid tumors like PDAC has relied on toxic systemic chemotherapy backbones (such as mFOLFIRINOX or gemcitabine plus nab-paclitaxel) that offer only modest survival extensions. Verastem’s VS-7375 directly addresses this therapeutic void as an oral, highly selective small-molecule inhibitor engineered to bind both the active and inactive states of the mutated KRAS G12D protein. By selectively turning off this aberrant signaling, VS-7375 is designed to disrupt the core oncogenic driver pathway, arresting cell-cycle progression and inducing apoptosis exclusively within mutant tumor cells while sparing wild-type tissue.
The open-label, multicenter Phase 1/2 dose-escalation and cohort-expansion study evaluated Verastem’s oral KRAS G12D inhibitor, VS-7375, in patients with advanced, refractory KRAS G12D-mutated solid tumors, focusing primary endpoints on safety, tolerability, and determining the recommended Phase 2 dose. While early data from an expansion cohort of 14 heavily pre-treated, second- or third-line (2-3L) metastatic pancreatic ductal adenocarcinoma (mPDAC) patients remained light on mature, radiological RECIST efficacy numbers, it demonstrated anti-tumor activity using a biomarker, with 93% (13 out of 14) of patients achieving a clinically significant ≥50% reduction in the prognostic tumor biomarker CA19-9. Additionally, VS-7375 proved to be generally well-tolerated with an on-target safety profile (such as manageable gastrointestinal issues and rash) what was free of treatment-limiting cumulative toxicities during the trial’s observation period. This biochemical response indicates that VS-7375 could be exerting therapeutic pressure on a notoriously lethal malignancy, potentially positioning the asset to address an unmet need left untouched by existing treatments.
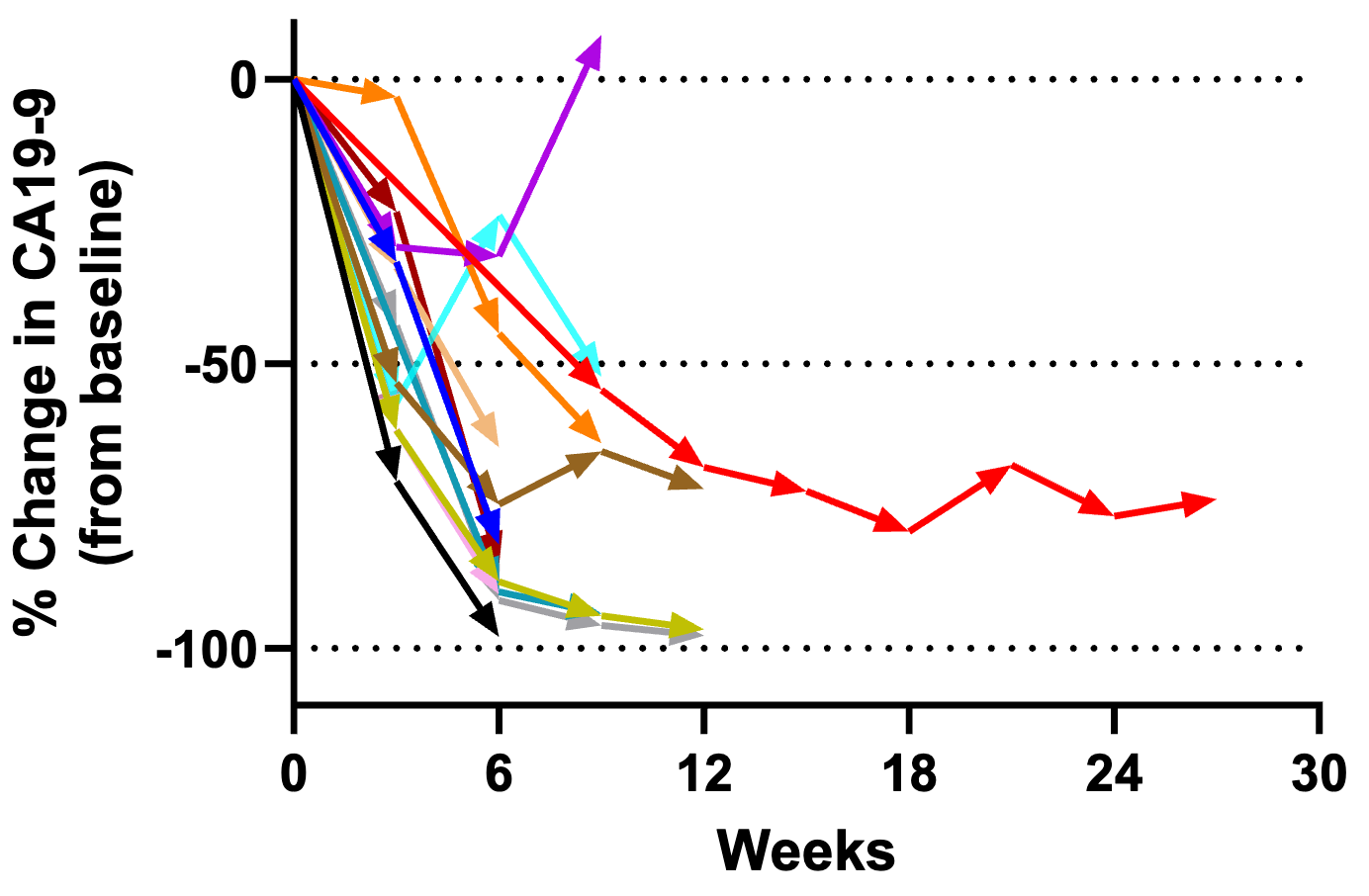
Rather than lingering in heavily pre-treated, refractory populations where tumors have accumulated bypass resistance mechanisms, Verastem is moving the asset forward into early-line treatment. Verastem announced plans to initiate a pivotal Phase 3 clinical trial evaluating VS-7375 in the first-line (1L) PDAC setting. The global Phase 3 study is scheduled to begin enrollment in the first half of 2027, likely evaluating VS-7375 in combination with standard frontline chemotherapy backbones (e.g., mFOLFOXIRINOX or gemcitabine/nab-paclitaxel).
Sources: Verastem press release, Verastem slide deck
Descriptive data releases without numerical data
Merck - Tulisokibart (TL1A inhibitor) / Phase 3 (UC): Merck has announced positive topline results from its Phase 3 ATLAS-UC induction-only study (Study 2) evaluating tulisokibart (MK-7240), an investigational humanized monoclonal antibody targeting TL1A to address immuno-fibrosis in adults with moderately to severely active ulcerative colitis (UC). The trial successfully met its primary endpoint of clinical remission, as measured by the Modified Mayo Score (MMS) at Week 12, marking the first successful Phase 3 victory for an anti-TL1A biologic. Tulisokibart also achieved key secondary endpoints, including endoscopic improvement, clinical response, and histologic-endoscopic mucosal improvement, while demonstrating a well-tolerated safety profile consistent with previous Phase 2 observations. Since Merck’s initial announcement was limited to qualitative topline conclusions without granular numerical data, this analysis relies strictly on the sponsor’s self-reported summaries pending full presentation at a future medical congress. Sources: Merck press release.
Love biotech? Check out Biotech Readout’s full content library, or navigate directly to a segment that interests you:
Frontiers in Medicine: Exploring the frontiers of our understanding and treatments for disease.
Medical History: Recovering forgotten relics in the history of medicine.
Acquisitions: Exploring the innovation behind acquired companies.
Weekly Readout: A digest of new clinical data from the past week.

To contact us, please send us an email at biotechreadout@gmail.com
Disclaimers
Investigational Status Disclaimer
The therapeutic candidates discussed in this newsletter are currently in clinical development and have not been approved for commercial sale by the U.S. Food and Drug Administration (FDA), the European Medicines Agency (EMA), or other global regulatory authorities. Their safety and efficacy have not been established. References to pipeline products and ongoing clinical trials involve significant risks and uncertainties. Statements regarding the potential safety, potency, or efficacy of investigational drugs reflect current hypotheses and are not a guarantee of future performance or regulatory clearance. The outcome of clinical trials is inherently unpredictable, and clinical results from earlier stages may not be predictive of results in later, larger-scale trials.
No Medical Advice Disclaimer
This newsletter is for informational and educational purposes only. The content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or another qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read in this publication.
No Patient-Provider Relationship Disclaimer
The information provided in this newsletter is for educational and analytical purposes only. Receipt of this information, or any interaction with this content, does not create a physician-patient, pharmacist-patient, or any other professional-provider relationship between you and the authors or publishers. This newsletter should not be used as a substitute for a personal consultation with a qualified healthcare professional.
Forward-Looking Statements Disclaimer
This newsletter contains “forward-looking statements” regarding future events, including clinical trial timing, regulatory milestones, and projected market performance. These statements are based on current expectations and assumptions that are subject to significant risks and uncertainties. Actual results may differ materially from those expressed or implied. We undertake no obligation to update these statements as a result of new information or future developments.
Third-Party Links & Content Disclaimer
This newsletter contains links to third-party websites, including clinical trial registries and corporate presentations. Biotech Readout does not endorse, guarantee, or assume responsibility for the accuracy or reliability of any information offered by third-party providers.
Errors and Omissions Disclaimer
While we strive for technical accuracy, the information in this newsletter is provided on an “as is” basis with no guarantees of completeness, accuracy, or timeliness. Biotech Readout assumes no liability for any errors or omissions in the content of this publication.
Non-Endorsement Disclaimer
Any reference to specific commercial products, processes, or services by trade name, trademark, or manufacturer does not constitute or imply an endorsement or recommendation by the author. All trademarks are the property of their respective owners.
No Investment Advice Disclaimer
This newsletter is for informational purposes only and does not constitute financial, investment, or legal advice. The author is not a registered investment advisor. You should consult with a professional financial advisor before making any investment decisions. The biotechnology sector is highly volatile; past performance is not indicative of future results.
Conflict of Interest Disclaimer
The author of this newsletter maintains a position of independence. At the time of publication, the author holds no direct financial interest, equity, or options in any of the companies mentioned in this report. No compensation has been received from any third party to feature or analyze specific therapeutic candidates or corporate entities.