
CAR-T, Part 3
How Dr. Georg Schett sparked renaissance in autoimmune disease

Disclaimer: This newsletter is for educational and informational purposes only and does not constitute medical, investment, or financial advice, nor does it establish a provider-patient relationship. Content may include forward-looking statements and discussions of investigational therapeutic candidates that are not FDA/EMA approved; their safety and efficacy remain unestablished and clinical outcomes are unpredictable. While we strive for accuracy, all information is provided as is without guarantees. This newsletter is independent, and the author holds no financial positions in the companies mentioned nor receives third-party compensation for this coverage. Please find a complete version of our disclaimers at the bottom of this article and on our About page.

Introduction
In Part 1 of this three-part Frontiers in Medicine series on CAR-T, we discussed the Immunology Civil War, the discovery of T-cells, and their rebirth as medicines. We followed that up with Part 2, where we discussed three therapeutic modalities that improved on the logistical and/or toxic pre-conditioning flaws of autologous CAR-T: allogeneic CAR-T, in vivo CAR-T, and T-cell engagers (TCE).
In Part 3, we pivot to something completely different, but still CAR-T. Could we apply this powerful modality to diseases outside of cancer? Here, we explore how one curious scientist asked that question and sparked a renaissance in autoimmune disease.
The Jewel of Innsbruck
Nestled deep in the heart of the Austrian Alps, Innsbruck is a rare gem where urban elegance seamlessly blends with rugged, high-altitude wilderness. Often called the “Capital of the Alps,” it offers a striking visual contrast: a fairytale historic center of pastel-colored buildings bordered by the rushing, glacial waters of the River Inn, all framed by the dramatic, jagged peaks of the Nordkette range that seem to rise directly out of the city streets.

Founded in 1669, the University of Innsbruck (Universität Innsbruck) is Austria’s leading hub of higher education. The intellectual roots of the university date back to 1562, well before its official founding. Ferdinand I, the Holy Roman Emperor, established a Jesuit college and grammar school in Innsbruck. This was the height of the Counter-Reformation in Europe. The Habsburg monarchy wanted a strong intellectual bastion in the Alps to counter the spread of Protestantism from the north.
By the late 19th century, the old campus structures were bursting at the seams. During this era, Innsbruck became a global focal point for groundbreaking scientific discovery, eventually yielding three separate Nobel Prizes:
Fritz Pregl (Chemistry, 1923): Developed groundbreaking methods for quantitative organic microanalysis while working in Innsbruck, which revolutionized the precise measurement of very small amounts of substances and significantly advanced chemical, biological, and medical research.
Adolf Windaus (Chemistry, 1928): Recognized for his research into sterols and their connection to vitamins (he studied in Innsbruck under Pregl).
Victor Franz Hess (Physics, 1936): Discovered cosmic radiation through daring high-altitude balloon flights, later heading the university’s Institute of Physics.
Like all Austrian institutions, the university suffered immense intellectual and moral damage during World War II. Numerous Jewish faculty members, independent scholars, and students were systematically expelled, and the university was temporarily renamed “Deutsche Alpenuniversität” before being liberated and restored at the end of World War II.
Today, the University of Innsbruck is universally recognized as a global juggernaut in quantum physics. Pioneered by world-renowned physicists like Anton Zeilinger (who conducted historic quantum teleportation experiments here before winning the 2022 Nobel Prize) and Rainer Blatt, the university’s labs routinely make international headlines, keeping this 350-year-old alpine institution at the cutting edge of human knowledge.

In the late 1980s, Georg Schett stepped into the lecture halls of the University of Innsbruck to begin his medical studies. As Schett moved through the grueling years of medical school, his focus began to narrow onto a specific, fascinating frontier: the human immune system. He wanted to understand the absolute molecular basics of why the body’s defenses occasionally malfunction. The sheer complexity of autoimmune reactions, where the body mistakenly launches an attack on its own structures, became his central obsession. By the time he successfully completed his doctoral thesis and graduated with his medical degree in 1994, he had resolved to cross the bridge from pure clinical practice into dedicated scientific discovery.
Rather than leaving the alpine valley immediately upon graduation, Dr. Schett chose to anchor his early professional steps in Innsbruck. He accepted a role as a research associate, stepping into the highly specialized laboratories of the Research Institute for Biomedical Aging Research, an elite arm of the Austrian Academy of Sciences located right in the city. This post-graduate window was a defining period of transition. Leaving behind the passive learning of a medical student, Dr. Schett assumed the active mantle of a laboratory scientist. He spent his days immersed in cell cultures, pipettes, and microscopic analysis. His primary scientific focus during these years was investigating the immunology of atherosclerosis (the hardening of the arteries) and antibody-mediated damage to endothelial cells.
By 1996, Dr. Schett’s foundational chapter in Innsbruck drew to a close. His meticulous work at the Research Institute had earned him recognition, including the prestigious Prize of the Austrian Society of Immunology and Allergology. Armed with the deep scientific training he had forged in the Alps, he left the valley for the nation’s capital, joining the University of Vienna to begin his clinical specialty training in internal medicine and rheumatology.
A Rheum with a View
After spending his early postgraduate years focused on laboratory research in aging and vascular immunology in Innsbruck, Dr. Schett recognized that unlocking the mysteries of autoimmune conditions required direct, comprehensive immersion in patient care. He joined the Department of Medicine at the University of Vienna to undertake his intensive postgraduate clinical training.
The environment in Vienna was fast-paced and highly demanding. Balancing late-night hospital rounds with early-morning laboratory assessments, Dr. Schett pursued a rigorous specialization in Internal Medicine. Working inside one of Europe’s largest medical epicenters, he encountered a massive diversity of complex systemic illnesses. This daily clinical exposure deepened his resolve to pinpoint exactly why the immune system turns against the body’s own tissues. He successfully completed his primary internal medicine board certification in 2001.
With his foundational residency complete, Dr. Schett advanced directly into his subspecialty training in Rheumatology, a medical speciality dedicated to diagnosing and treating these autoimmune and musculoskeletal conditions. He focused his scientific and clinical energy heavily on the molecular mechanisms driving severe, chronic joint and tissue inflammation. During this period, he maintained a dual identity as both a practicing clinician and an active investigator. He sought to bridge the gap between bench and bedside by examining the immunology of atherosclerosis and investigating the exact nature of antibody-mediated endothelial cell damage.
In 2002, Dr. Schett’s innovative research proposals caught the attention of the Austrian science community, earning him the prestigious START Award from the Austrian Science Fund. This highly competitive grant provided substantial independent funding, allowing him to establish his very first dedicated research group specifically focused on the pathogenesis of arthritis in Vienna.
Equipped with his own laboratory and a growing team of young scientists, Dr. Schett’s academic output accelerated dramatically. His work untangling the complex inflammatory pathways of lupus and rheumatoid arthritis yielded significant publications and elevated his status within the university. In 2003, his scientific and teaching contributions culminated in his formal academic habilitation, and he was promoted to Professor of Internal Medicine at the university. In 2004, he finalized his subspecialty requirements, officially certifying as a Specialist Physician in Rheumatology and advancing to a senior physician role within the hospital.
By the time his decade-long chapter in Vienna drew to a close, Dr. Schett had evolved from a trainee into an influential academic leader. The clinical acumen he developed on the wards of Vienna, combined with the administrative and scientific freedom granted by his 2002 START Award, provided a springboard for his future endeavors. When he eventually departed for Germany in 2006 to accept the Chair of Internal Medicine 3 at FAU Erlangen-Nürnberg, he carried with him the deep clinical insights forged at the University of Vienna, insights that would later enable him to uncover something most unexpected. Before we get to Dr. Schett’s pivotal work, we’re going to take a detour into the history of lupus.
Wolves and Lesions
The history of Systemic Lupus Erythematosus (SLE) is a fascinating scientific journey. For centuries, physicians only knew lupus by its outward manifestations on the skin. Due the disease’s often destructive, ulcerating facial lesions that erode tissue, medieval doctors thought the damage resembled the devastating injuries left by a wolf attack. The term lupus (the Latin word for “wolf”) was first applied to medical skin conditions in the 12th century by a prominent Italian physician named Dr. Rogerius Frugardi, who used it to describe severe, gnawing facial ulcers.
In 1845, the French dermatologist Dr. Ferdinand von Hebra made a critical breakthrough by linking these disparate skin conditions together. He mapped out the distinct, symmetric facial redness that stretched across the nose and cheeks. Due to its vivid red color, he appended the Greek-derived word erythematosus (red/flushed). Dr. Von Hebra was also among the first to formally describe it as a “butterfly-shaped” pattern.

Until the late 19th century, lupus was viewed strictly as a dermatological problem. That changed entirely due to the brilliant observations of an Austrian dermatologist, Dr. Moriz Kaposi (famous also for describing Kaposi’s sarcoma). In 1872, Dr. Kaposi realized that some patients with the characteristic butterfly rash also suffered from fevers, severe weight loss, joint arthritis, and fatal internal organ failure particularly in the lungs and kidneys. He proposed that lupus was actually two diseases: a localized skin version (discoid lupus) and a systemic version that attacked the inside of the body. With Kaposi’s discovery, the modern concept of Systemic Lupus Erythematosus (SLE) was born.
In the early 1900s, legendary clinician Sir William Osler expanded on Kaposi’s work. He mapped the systemic nature of SLE in detail, noting that the disease could flare and recede, quietly damaging the heart, kidneys, and central nervous system even when the skin appeared completely clear. The mid-20th century transformed lupus from a descriptive clinical puzzle into a molecularly defined syndrome through a sequence of laboratory breakthroughs:
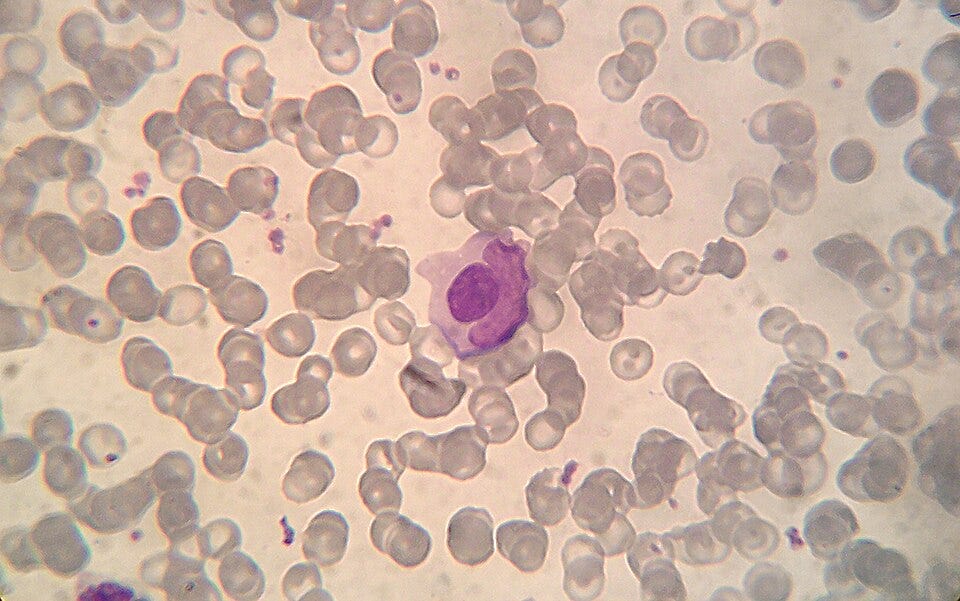
Discovery of the LE Cell (1948): Dr. Malcolm Hargraves discovers the “Lupus Erythematosus (LE) cell” in bone marrow plasma. This is the first definitive diagnostic laboratory marker for the disease, showing that something in the blood was causing white blood cells to devour dead cellular material. Today, we recognize an LE cell as a neutrophil (or sometimes a macrophage) that has ingested the denatured nuclear material of another damaged cell. A SLE patient’s serum can contains autoantibodies, originally called the LE factor, which are specifically anti-histone and anti-DNP (deoxyribonucleoprotein) antibodies. These autoantibodies attach to the newly exposed, naked nucleus of the damaged cell. The binding of these autoantibodies denatures the nuclear material, transforming it into a smooth, purple-staining, homogeneous sphere known as a hematoxylin body or an LE body.
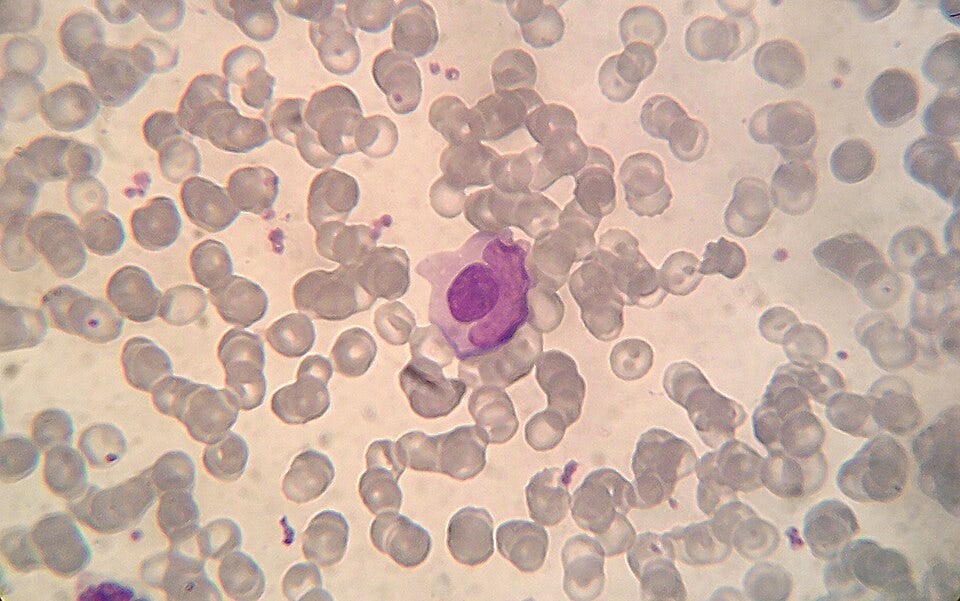
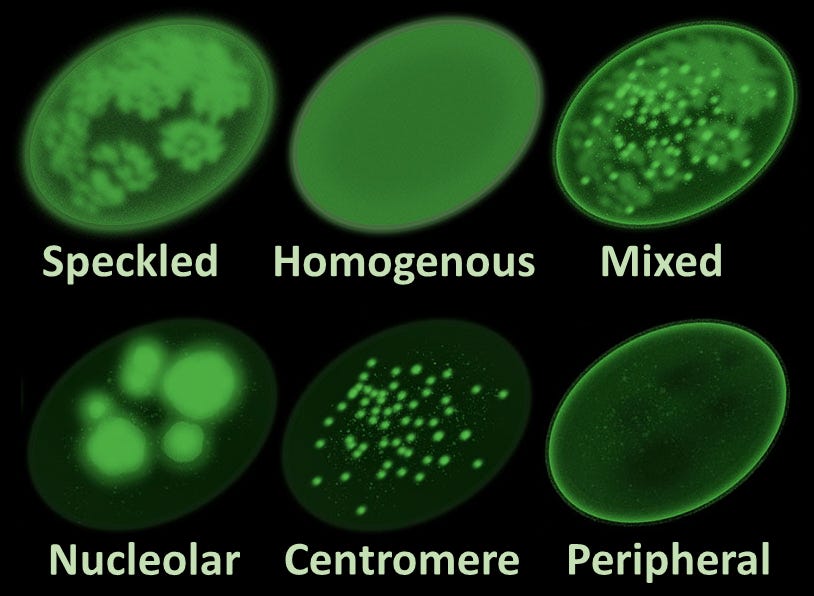
Microphotograph of a LE cell, May Grünwald Giemsa stain, 100x immersion objective + 3:1 digital ampliation; Source: Wikimedia The Antinuclear Antibody (ANA) Test (1957): Researchers develop fluorescent antibody staining techniques, leading to the invention of the ANA test. This proves conclusively that lupus is an autoimmune disease, the body’s immune system is generating antibodies that mistakenly target the nuclei of its own healthy cells.
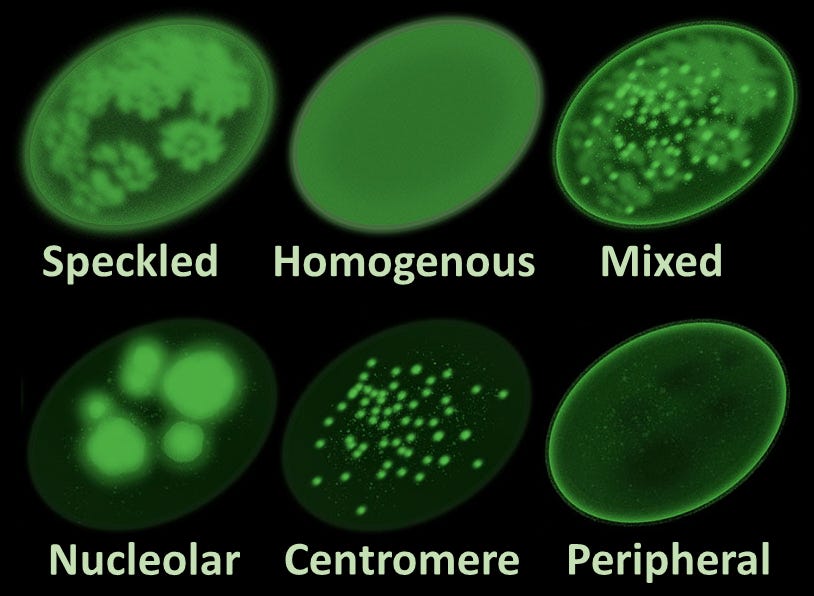
Main antinuclear antibody patterns on immunofluorescence; Source: Al-Mughales JA. Minor edits by Mikael Häggström, M.D. Author info - Reusing images- Conflicts of interest: NoneMikael Häggström, M.D., CC BY 4.0 <https://creativecommons.org/licenses/by/4.0>, via Wikimedia Commons Discovery of Anti-dsDNA and Anti-Sm Autoantibodies (1960s): Scientists isolate highly specific autoantibodies, such as anti-double-stranded DNA (anti-dsDNA) and Anti-Smith (Anti-Sm). These markers allow clinicians to distinguish SLE from other rheumatological conditions like rheumatoid arthritis with extreme accuracy.
{kind=link}
{kind=link}
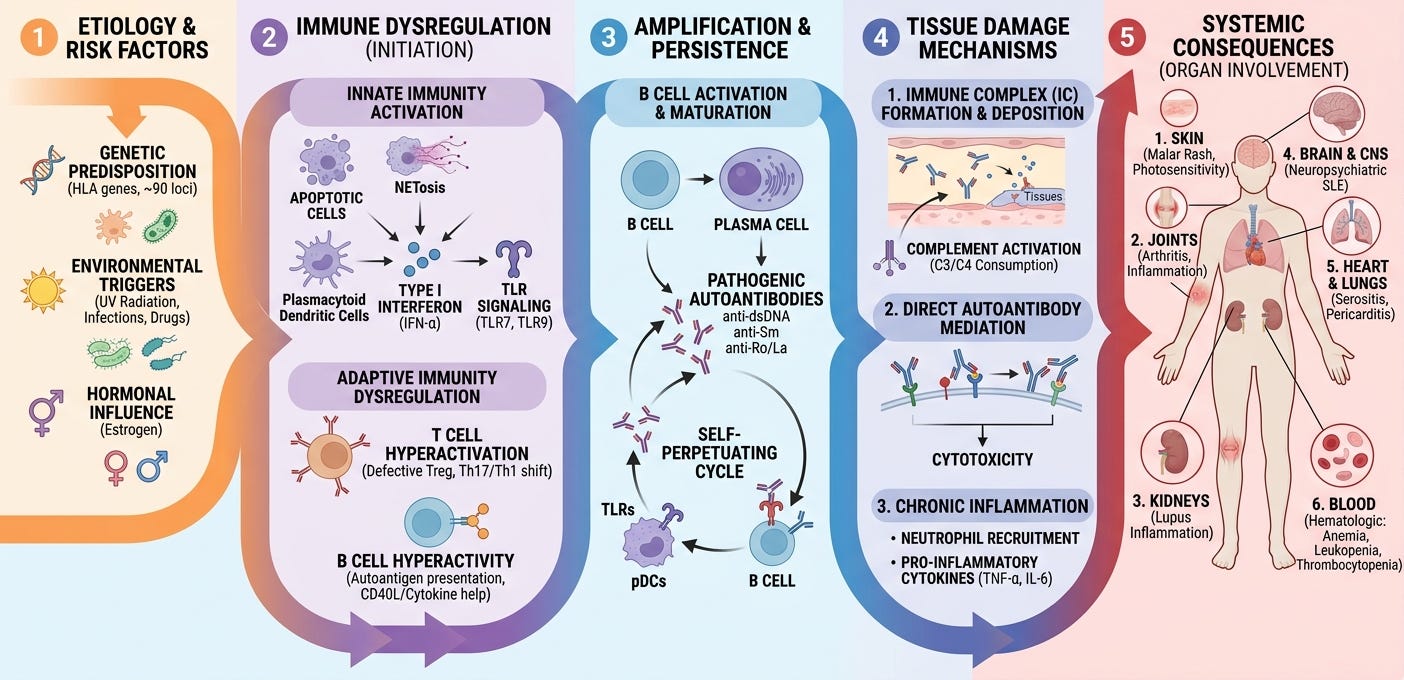
The modern pathophysiological understanding of Systemic Lupus Erythematosus (SLE) views the disease not as a single linear malfunction, but as a complex, multi-step cascade. It is characterized by a profound failure in how the body clears cellular debris, leading to a perpetual cycle of autoantibody production, chronic immune complex deposition, and widespread tissue damage.
SLE begins at the intersection of a vulnerable genome and environmental insults. Genome-wide association studies (GWAS) have identified over 100 susceptibility loci. These genes predominantly regulate immune pathways, such as interferon signaling (e.g., IRF5, TYK2), immune complex clearance (e.g., FCGR alleles, early complement components like C1q, C2, C4), and B/T-cell activation thresholds. External factors act as the initial kinetic catalysts. Ultraviolet (UV) radiation damages keratinocytes, inducing widespread cellular apoptosis (programmed cell death). Other triggers include viral infections (particularly Epstein-Barr Virus / EBV through molecular mimicry), hormonal fluctuations (estrogen enhances lymphocyte activation, explaining the 9:1 female-to-male bias), and certain medications or toxins.
In a healthy individual, billions of cells undergo apoptosis daily. The immune system efficiently clears this cellular debris without causing inflammation. In SLE, this waste-disposal mechanism breaks down entirely. Macrophage phagocytosis is sluggish, and genetic deficiencies in complement proteins mean apoptotic cells linger in tissues. As these dying cells undergo secondary necrosis, they burst open, spilling intracellular contents, including double-stranded DNA (dsDNA), histones, and ribonucleoproteins (such as Smith/Sm, Ro, and La), into the extracellular space. These normally hidden components are now exposed to the immune system as necroantigens. Once these nuclear antigens are exposed, the immune system treats them as dangerous foreign invaders, triggering a self-amplifying loop:
The Innate Response & Type I Interferon Hyperactivation: Plasmacytoid dendritic cells (pDCs) recognize the spilled nucleic acids via endosomal receptors known as Toll-Like Receptors (TLR7 and TLR9). In response, pDCs pump out massive amounts of Type I Interferon-alpha (IFN-alpha). This creates a perpetual “interferon signature” that hyper-activates surrounding immune cells, mimicking a chronic, unending viral infection.
The Adaptive Response (B and T-Cell Dysregulation): Driven by the interferon storm, helper T-cells activate self-reactive B-cells. This survival loop is further fueled by elevated levels of cytokines like BAFF/BLyS (B-cell activating factor). Instead of producing normal antibodies, these dysregulated B-cells mature into plasma cells that churn out an arsenal of Antinuclear Antibodies (ANAs) targeting the body’s own dsDNA, histones, and ribonucleoproteins.
The final, clinical phase of SLE occurs when the newly minted autoantibodies bind to the freely floating nuclear antigens, creating structural lattices known as immune complexes. These heavy immune complexes travel through the bloodstream and lodge physically within the narrow basement membranes of vital organs:
Kidneys (Lupus Nephritis): Complexes trap inside the renal glomeruli.
Skin: Deposition occurs at the dermo-epidermal junction (leading to the malar/butterfly rash).
Blood Vessels & Joints: Deposition in vessel walls (vasculitis) and synovium (arthritis).
Once trapped, these complexes fix and activate the complement cascade (generating anaphylatoxins like C3a and C5a). This acts as a chemical beacon, drawing in neutrophils and macrophages. These inflammatory cells release destructive lysosomal enzymes and reactive oxygen species, causing chronic scarring, tissue necrosis, and eventual organ failure.
Dousing Flames with Kerosene
Before the mid-20th century, the biological underpinnings of lupus were completely unknown, and a diagnosis of systemic lupus was frequently a terminal event. Treatment was largely experimental, symptomatic, and highly toxic. In the late 19th and early 20th centuries, dermatologists used injections of heavy metals, specifically arsenic (in the form of Fowler’s solution) and gold salts, to treat the severe skin lesions of lupus. While gold salts provided mild anti-inflammatory relief, they frequently caused severe kidney and bone marrow toxicity.

In 1894, British physician Dr. J.F. Payne discovered that quinine (an antimalarial drug) significantly improved lupus skin rashes. This laid the early foundation for what would eventually become a modern cornerstone of lupus management. The modern therapeutic era was born on September 21, 1948, when Mayo Clinic clinicians first administered “Compound E” (cortisone) to a patient with rheumatoid arthritis. By the early 1950s, the mass production of synthetic prednisone completely transformed the prognosis for acute SLE. For the first time in medical history, physicians possessed a fast-acting, blunt-force instrument capable of staving off life-threatening inflammatory flares and reducing the risk of acute renal or pulmonary failure. However, this treatment carried a heavy price. High-dose, long-term steroid use introduced severe secondary complications, including osteoporosis, avascular necrosis, severe infections, and accelerated cardiovascular disease.
As scientists recognized that lupus was driven by an overactive, autoantibody-producing immune system, they began borrowing chemotherapy drugs from oncology to aggressively suppress lymphocyte proliferation. An improved derivative of chloroquine, sold as Plaquenil (1955; hydroxychloroquine), was approved. It gradually became recognized not just as a skin treatment, but as a foundational life insurance policy for all lupus patients, shown to reduce disease flares and protect organ systems.
Drugs like azathioprine (1960s), cyclophosphamide (1970s), and later mycophenolate mofetil (MMF) in the 1990s became the standard of care for severe complications like lupus nephritis. Cyclophosphamide, administered in heavy monthly intravenous pulses, successfully kept patients off dialysis but carried profound side effects, including bone marrow suppression and permanent infertility.
For half a century, treating systemic lupus erythematosus (SLE) meant choosing between the blunt-force anti-inflammatory effects of corticosteroids or the scorched-earth cytotoxicity of oncology-derived immunosuppressants like cyclophosphamide. By the early 2000s, advances in molecular immunology allowed scientists to move away from systemic destruction and toward targeted therapy. Instead of shutting down the entire immune system, engineered monoclonal antibodies were designed to intercept specific cytokines, surface receptors, and survival signals essential to the lupus autoimmune cascade. The story of this era is defined by hard-fought clinical trials, historic failures that reshaped study designs, and resulted in a handful of landmark molecular triumphs.
Reducing Heat to a Simmer
Since pathogenic autoantibodies (like anti-dsDNA) are the primary executioners of tissue damage in SLE, the earliest logical targets of the biologic era were B-cells, the cells responsible for producing those antibodies.
In the early 2000s, clinicians began borrowing Rituxan (rituximab), a chimeric monoclonal antibody that targets the CD20 antigen expressed on the surface of pre-B and mature B-lymphocytes. In open-label, real-world clinical settings, rituximab effect were described as profound. Patients with severe, treatment-resistant lupus nephritis or central nervous system disease experienced dramatic, rapid turnarounds. However, when put to the test in large, rigorous randomized controlled trials, specifically the EXPLORER trial (2010) for extra-renal lupus and the LUNAR trial (2012) for lupus nephritis, rituximab failed to meet its primary endpoints. Some physicians maintain that failure of rituximab for SLE was a failure of early trial design rather than a lack of biological activity. Patients in both the drug and placebo arms were allowed to take heavy background doses of high-dose corticosteroids and immunosuppressants (like mycophenolate mofetil). Critics suggest that this heavy background therapy effectively masked the clinical efficacy of rituximab, making it statistically impossible to show a distinct benefit.
Drug developers pivoted from completely destroying B-cells to subtly altering the microenvironment that keeps them alive. This led to the targeting of BAFF (B-cell activating factor, also known as BLyS, B-lymphocyte stimulator). BLyS is a naturally occurring cytokine that acts as a vital survival signal for B-cells as they transition from immature cells into mature, antibody-producing plasma cells. In SLE patients, BLyS levels are chronically elevated, effectively keeping self-reactive, autoreactive B-cells alive when they should naturally be cleared by apoptosis.
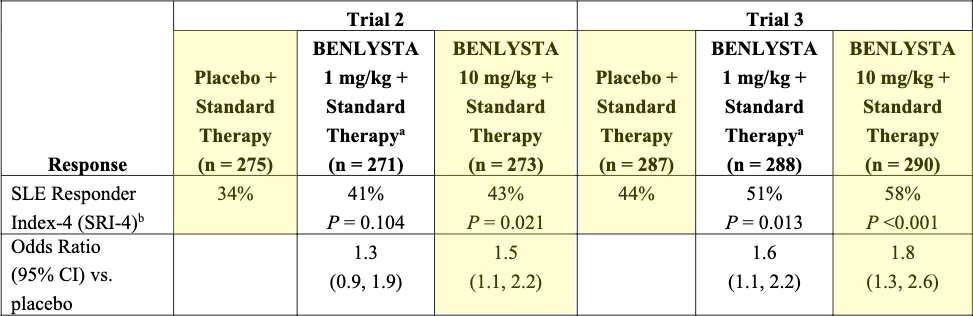
In 2011, Benlysta (belimumab), a fully human IgG1 monoclonal antibody that binds to and neutralizes soluble BLyS, made history as the first-ever biologic agent approved by the FDA specifically for SLE. Learning from the rituximab failures, the pivotal BLISS-52 and BLISS-76 Phase 3 trials utilized a newly designed, comprehensive composite endpoint called the SLE Responder Index (SRI), a composite clinical trial endpoint that measures meaningful global improvement in:
Lupus activity (via a ≥4-point reduction in the SELENA-SLEDAI score)
While strictly requiring that the patient experiences no new severe organ flares (via the BILAG index)
And no overall clinical deterioration (via the Physician Global Assessment)
Benlysta demonstrated a statistically significant reduction in disease activity, lowered the frequency of severe flares, and allowed clinicians to successfully taper patients off toxic background doses of prednisone. In 2020, its approval was expanded to include Lupus Nephritis based on the successful BLISS-LN trial.
While Benlysta successfully managed the adaptive immune response (B-cells), the modern pathophysiological understanding of lupus highlighted an upstream culprit in the innate immune system: the Type I Interferon (alpha, beta, and kappa) pathway. Over 60-80% of adult SLE patients exhibit a profound upregulation of genes driven by Type I interferons, a phenomenon known as the Type I Interferon Signature. This persistent interferon storm constantly activates dendritic cells, drives T-cell differentiation, and pushes B-cells to produce autoantibodies, tricking the body into thinking it is fighting a perpetual viral infection.
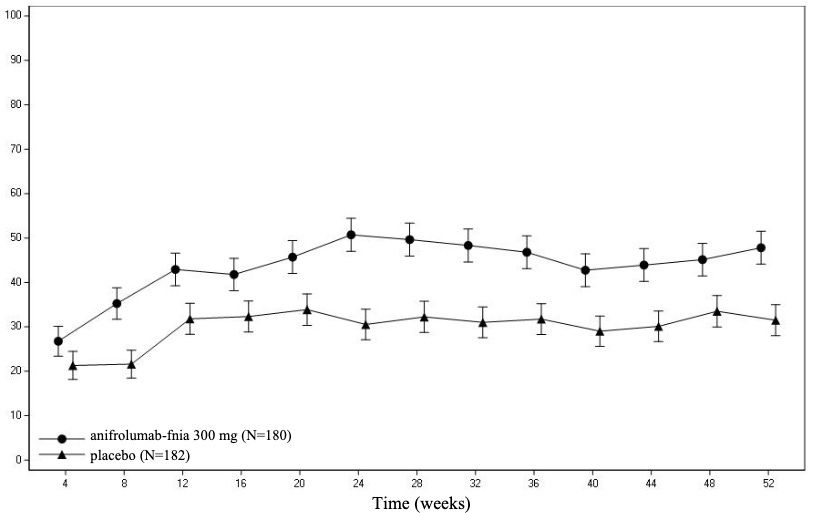
Early attempts to neutralize individual interferon-alpha molecules directly (such as sifalimumab and rontalizumab) yielded disappointing clinical results because the body simply compensated by utilizing other interferon subtypes. Scientists changed the target from the floating cytokines to the actual lock they all must open: the Type I Interferon Receptor Subunit 1 (IFNAR1). In August 2021, the FDA approved Saphnelo (anifrolumab), a fully human monoclonal antibody that binds directly to IFNAR1, completely blocking the signaling of all Type I interferons. Though its initial TULIP-1 trial missed its primary endpoint due to rigid score parameters, the subsequent TULIP-2 trial utilized the BICLA (British Isles Lupus Assessment Group-based Composite Lupus Assessment) index. Saphnelo demonstrated significant clinical efficacy, showing rapid and sustained improvements in severe skin disease (cutaneous lupus) and joint arthritis, while drastically reducing the patient’s dependence on oral corticosteroids.
The emergence of Benlysta and Saphnelo reshaped the standard of care for lupus in a way that controlled the disease with molecular nuance rather than systemic devastation. However, even the most successful biologics of this era remained maintenance therapies, temporarily intercepting the inflammatory cascade. If a patient stops taking their monthly infusions of Benlysta or Saphnelo, the underlying molecular machinery eventually spins back up, and the disease returns. This brings us back to Dr. Georg Schett, who had departed from University of Vienna and arrived at the FAU Erlangen-Nürnberg in Germany.
Extinguishing Flames
When Dr. Georg Schett arrived at Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU) in 2006 to assume the prestigious Chair of Internal Medicine 3 (Rheumatology and Immunology), he inherited a department steeped in a long tradition of clinical excellence. Yet, he brought with him a distinct, forward-looking ambition honed during his years in Vienna and the United States: a determination to transition rheumatology away from lifelong disease management and toward definitive molecular solutions.
For more than a decade in Erlangen, Dr. Schett and his team pushed the boundaries of targeted immunology. They optimized the use of monoclonal antibodies and small-molecule inhibitors, but by late 2019, a sobering clinical picture persisted. In the hospital wards, a subset of patients with severe, refractory Systemic Lupus Erythematosus (SLE) were running out of options. These patients, often young women, presented with fulminant disease. Their bodies were locked in a hyper-accelerated autoimmune cascade, producing torrents of destructive autoantibodies that relentlessly attacked their kidneys, lungs, and central nervous systems. Even the most aggressive combinations of high-dose steroids, mycophenolate mofetil, and rituximab failed to stem the tide. Faced with these life-threatening impasses, Dr. Schett recognized that incremental maintenance therapies had hit a wall. To save these patients, the team needed to find a way to wipe out the autoreactive immunological memory. They needed an immune reset.
The conceptual breakthrough did not originate within traditional rheumatology literature. Instead, it came from looking across the medical aisle at oncology. For several years, hematology-oncology specialists had been achieving remissions in patients with terminal B-cell leukemias and lymphomas using Chimeric Antigen Receptor T-cell (CAR-T) therapy. This therapy involved extracting a patient’s own autologous T-cells and genetically engineering them in a lab to express a synthetic receptor targeting CD19, a lineage-specific surface protein found exclusively on B-lymphocytes. Once re-infused, these living cellular assassins hunted down and obliterated nearly every CD19-bearing cell in the body.
Dr. Schett made a profound, cross-disciplinary connection. The exact same cell type driving hematological malignancies, the B-cell, was the upstream engine driving the horrors of lupus. In SLE, dysregulated B-cells mature into plasma cells that continuously churn out antinuclear antibodies (ANAs) and anti-dsDNA, which form the tissue-destructive immune complexes. While traditional monoclonal antibodies like rituximab merely cleared B-cells from peripheral blood, they left deep-tissue sanctuaries (like the spleen, lymph nodes, and bone marrow) completely untouched. Rogue B-cell clones hid in these niches, quietly surviving to drive subsequent, devastating clinical flares. Dr. Schett hypothesized that engineered CD19 CAR T-cells, with their active tissue-homing mechanisms and robust proliferative capacity, could wipe out these rogue B-cells. They could penetrate the bone marrow niches, track down pathogenic B-cell clones, and potentially erase their autoimmune memory.
In early 2021, a 20-year-old female patient arrived at the Erlangen university clinic in critical condition. Her lupus was in a state of catastrophic acceleration: she was suffering from severe, rapidly progressing lupus nephritis (kidney failure), active arthritis, and profound endocardial heart inflammation. Every standard and experimental immunosuppressive regimen had failed. Under strict compassionate-use guidelines and in close collaboration with FAU’s hematology-oncology teams, Dr. Schett prepared to administer the world’s first-ever CAR T-cell infusion for an autoimmune disease.
The clinical team held its breath. In oncology, CD19 CAR T-cell therapy is notoriously high-risk, frequently triggering severe Cytokine Release Syndrome (CRS), a violent, systemic inflammatory storm caused by T-cells rapidly engaging with billions of circulating tumor cells. However, as the engineered cells were slowly infused back into the young woman’s bloodstream, Dr. Schett’s team observed a profound, highly encouraging divergence from the oncology experience. Since the baseline target burden of B-cells in an autoimmune patient is orders of magnitude lower than a massive tumor load, the massive systemic cytokine spikes rarely materialized. The patient tolerated the infusion remarkably well, experiencing only a transient, mild, easily manageable low-grade fever. Within days, the engineered T-cells executed their molecular mission with absolute precision. In the peripheral blood and deep tissue biopsies, her circulating B-cells dropped to undetectable levels. The pathogenic architecture had been dismantled.
Dr. Schett and his Erlangen colleagues published these stunning initial single-patient results in NEJM in late 2021, quickly expanding the cohort to five patients in a 2022 Nature Medicine publication, all of whom mirrored a similar trajectory of drug-free clinical remission. To investigate whether this was a universal therapeutic principle rather than an isolated fluke of lupus biology, Dr. Schett expanded the clinical protocol at FAU to target other devastating, B-cell-driven autoimmune conditions. In early 2024, the team published a landmark, comprehensive case series in the NEJM tracking 15 patients across three distinct, severe autoimmune pathologies:
8 patients with Systemic Lupus Erythematosus (SLE)
3 patients with Idiopathic Inflammatory Myositis (severe muscle-destroying autoimmunity)
4 patients with Systemic Sclerosis / Scleroderma (fatal, widespread tissue fibrosis)
Every single one of the 15 treated patients achieved complete clinical remission according to strict international disease activity indexes. Every patient successfully discontinued all daily oral corticosteroids, background chemotherapies, and maintenance biologics. Long-term follow-up tracking patients past the two-year mark demonstrated that even after healthy, naive B-cells completely repopulated the body, the autoimmune diseases showed no signs of returning.
Crossing the Rheubicon
Dr. Georg Schett’s relationships with two biotech companies, Cabaletta Bio and Kyverna Therapeutics, show how paradigm-shifting academic research can cross into industry pipelines. Since his clinical trials at Erlangen were the first to prove that CD19-targeting CAR T-cells could successfully achieve an immune reset in refractory autoimmune patients, both companies aggressively sought his expertise to shape their clinical strategies. While Dr. Schett sits on the Scientific Advisory Board (SAB) for both, his structural engagements differ slightly in how they utilize his clinical datasets.
Before Dr. Schett’s results in autoimmune disease, Cabaletta’s entire platform was built on CAART (Chimeric Autoantibody Receptor T cells). Traditional oncology CAR T-cells are broad bludgeons, they wipe out all B-cells (both healthy and diseased), but Cabaletta believed this was too dangerous and immunosuppressive for non-cancer patients. They engineered T-cells to display specific autoantigens (like DSG3 for Pemphigus Vulgaris or MuSK for Myasthenia Gravis). The goal was to target only the specific, rogue B-cells causing that exact disease, leaving the rest of the patient’s healthy B-cell immune system completely untouched. While elegant, this approach was highly niche, technically difficult to scale, and limited to diseases with perfectly mapped, single-antigen targets.
When Dr. Schett showed that a complete, transient depletion of all B-cells was not only safe but actually allowed the immune system to grow back healthy and naive, Cabaletta realized their hyper-targeted approach wasn’t necessary to cure these diseases. In late 2022, Cabaletta executed a rapid pivot. They launched their CARTA (Chimeric Antigen Receptor T cells for Autoimmunity) strategy, licensed a fully human CD19 binder from IASO Bio, and engineered CABA-201 (also called resecabtagene autoleucel or rese-cel for short) specifically choosing a 4-1BB co-stimulatory domain to match the exact structural layout Dr. Schett used in Erlangen. Rese-cel is now the center of gravity for Cabaletta. Their original CAART programs were put on the back burner while they rapidly pushed rese-cel into parallel Phase 1/2 trials (the RESET trials) across multiple indications like Lupus, Myositis, and Systemic Sclerosis, aiming for registrational readouts.
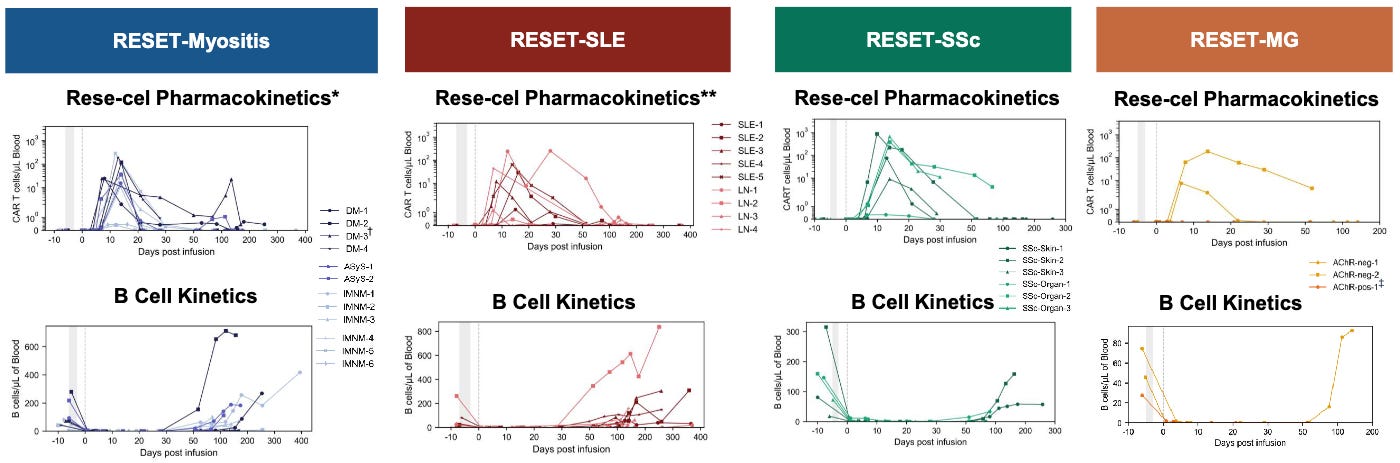
While Dr. Schett serves as a key member of Cabaletta’s SAB alongside pioneers like Dr. Carl June, his relationship goes a step further. Under an exclusive translational agreement struck in late 2022, Cabaletta’s scientists work directly with Dr. Schett’s academic team in Germany. The goal is to pool data to map out exactly how deep tissue B-cell depletion occurs, track how naive B-cells safely repopulate without autoantibodies, and isolate the exact immunologic biomarkers that predict a durable, drug-free remission. Dr. Schett’s real-world safety data, which notably showed very manageable Grade 1 Cytokine Release Syndrome (CRS) and zero Immune Effector Cell-Associated Neurotoxicity Syndrome (ICANS) cases, directly informed the design, preconditioning regimens, and initial dosing strategies for Cabaletta’s Phase 1/2 trials in lupus nephritis, systemic sclerosis, and myositis.
Kyverna underwent a similar pivot. Prior to Dr. Schett’s findings, Kyverna was focused on suppressing inflammation via Tregs (Regulatory T-cells) to damp down overactive immune responses and re-establish homeostasis rather than cytotoxically killing cells. They were building complex logic-gated circuits to quiet down tissue-specific autoimmune inflammation without killing the host cells. While they knew CD19 depletion had potential, they lacked an optimized construct for it.
When Dr. Schett’s early data validated that CD19-targeted cell death was the fastest known path to a clinical remission, Kyverna aggressively accelerated their B-cell depletion timeline. Instead of building a construct from scratch like Cabaletta, Kyverna went to the National Institutes of Health (NIH) and secured an exclusive, worldwide license for a unique, fully human anti-CD19 CAR construct. This specific construct (now KYV-101) had already been tested by the NIH in a 20-patient oncology trial and was prized for causing significantly lower levels of inflammatory cytokines.
Kyverna brought Dr. Schett onto their advisory board to help them position KYV-101 as the “safety-optimized” alternative. Armed with Dr. Schett’s blueprint on autoimmune cell behavior, Kyverna became the first of the two to secure FDA Investigational New Drug (IND) clearance for Lupus Nephritis in late 2022. Kyverna has leaned heavily into neuroimmunology. They are currently leading the regulatory race, having initiated a rolling Biologics License Application (BLA) submission for KYV-101 (miv-cel) in Stiff-Person Syndrome (SPS), making them the first company to bring an autoimmune CAR T therapy to the doorstep of FDA review.
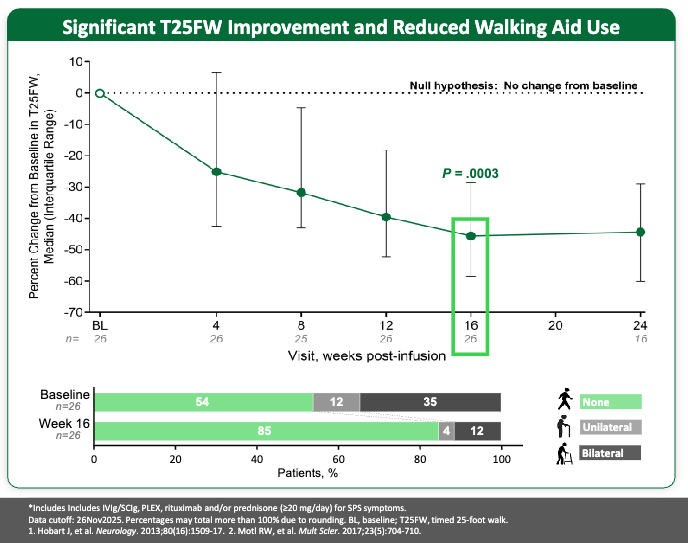
Engaged to be Married
As we mentioned in Part 2 of this series, T-cell engagers (TCE) provide an alternative to the logistical hurdles and toxic preconditioning required for autologous CAR-T cells, a benefit that holds true in autoimmune diseases. Following in the footsteps of their cellular counterparts, the core papers outlined the foundational proof-of-concept for TCEs in this setting.
The early, definitive clinical translation of this modality into rheumatology was highlighted by a key 2024 study in Nature Medicine, once again led by Dr. Georg Schett’s team. They tested a molecule featuring binding arms for CD3 on the surface of the patient’s native cytotoxic T-cells and CD19 on B-lymphocytes. By physically “marrying” the two cells, the TCE triggered the T-cell to release perforins and granzymes, destroying the B-cell. The paper showed that this off-the-shelf approach could penetrate deep tissues and achieve a profound clearance of pathogenic B-cells in severe, refractory rheumatoid arthritis (RA), mirroring the early success of CAR T-cell therapy.
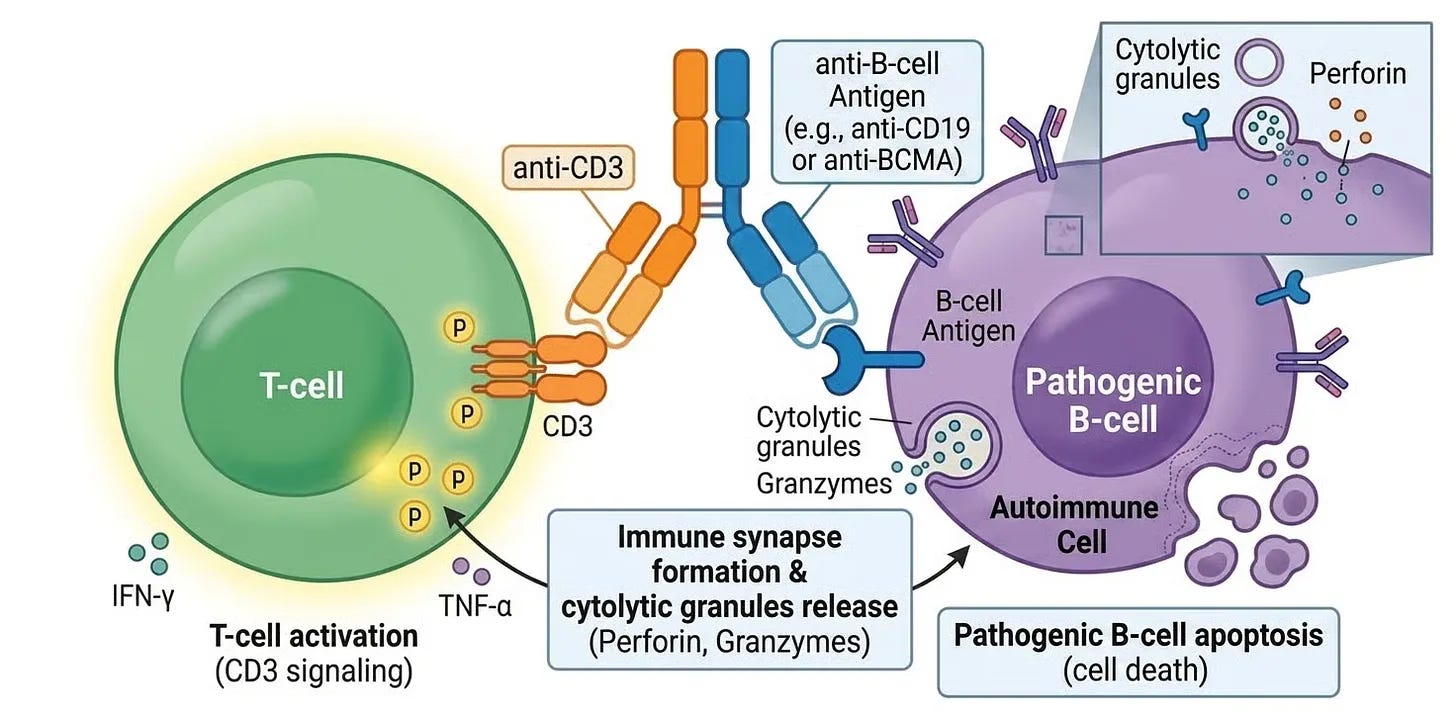
While CD19-directed therapies successfully wipe out B-cells and short-lived plasmablasts, they consistently hit a biological wall: long-lived plasma cells do not express CD19. In many severe autoimmune diseases (such as systemic sclerosis or Sjögren’s syndrome), the true drivers of the disease are mature, long-lived plasma cells hidden deep within the bone marrow. These cells continuously pump out tissue-destructive autoantibodies (like SS-A/Ro or anti-Scl-70) and can survive both anti-CD20 monoclonal antibodies (rituximab) and CD19 CAR T-cells. To bridge this gap, Dr. Schett’s clinic shifted focus to a different target: B-Cell Maturation Antigen (BCMA), which is highly expressed on mature plasma cells and plasmablasts. They published their breakthrough clinical results in NEJM. They saw:
Rapid, Complete Drug-Free Remission: Across treated patients with severe systemic lupus erythematosus (SLE), primary Sjögren’s syndrome, idiopathic inflammatory myositis, and systemic sclerosis, disease activity scores plummeted rapidly. Patients completely discontinued all daily oral corticosteroids and background immunosuppressants.
Deep Plasma Cell and Tissue Clearance: The therapy achieved an swift depletion of peripheral B-cells and a vast eradication of deep bone marrow plasma cells, resulting in a marked drop in serum free light chains and the unprecedented clearing of entrenched autoantibodies.
Resolution of Active Inflammation: Functional imaging (such as 68Ga-FAPI-PET-CT scans) documented the objective resolution of deep tissue inflammation, such as active arthritis in the hands and knees, alongside sharp reductions in severe skin ulcerations and improvements in lung diffusion capacity.
While there are no TCEs currently approved specifically for autoimmune disease, several oncology-approved and novel engineered TCEs are progressing rapidly through Phase 1 and Phase 2 clinical evaluation. The clinical landscape spans three primary target strategies aimed at deeply depleting pathogenic B-cell and plasma-cell niches:
CD19 Targeting TCEs: These are the most common target for pan-B-cell depletion, reaching early pre-B cells up to mature B cells (while sparing long-lived plasma cells).
BCMA Targeting TCEs: These allow therapies to eliminate mature plasma cells and plasmablasts, which are responsible for secreting autoantibodies and are often missed by anti-CD20 or anti-CD19 agents.
Multi-Targeted TCEs: These have overlapping mechanisms and include:
CD20 x CD8 x CD3 TCEs (AstraZeneca’s AZD5492)
CD19 x BCMA x CD3 TCEs (Sanofi’s KT501, UCB/Candid’s CND460, Earandil’s HXN-1031)
CD19 x CD20 x CD3 TCEs (GSK’s GSK5926371, UCB/Candid’s CND319, Kali’s KT501)
Unlike oncology, where high-grade toxicities are tolerated for survival benefits, autoimmune applications require a vastly superior therapeutic window. These newer clinical-stage candidates prioritize:
Mitigating CRS and ICANS: Utilizing low-affinity CD3 arms, conditional masking, or slow subcutaneous absorption to flatten the cytokine peak.
Tissue-Level Depletion: Ensuring the bispecific effectively penetrates secondary lymphoid organs and inflamed tissues (e.g., the synovium or kidneys) to eliminate protected autoreactive niches that traditional monoclonal antibodies like rituximab fail to clear.
Conclusion
The multi-part journey of the CAR-T modality is a testament to the unpredictable, non-linear nature of scientific innovation. A platform built entirely to combat terminal blood cancers unexpectedly led to a renaissance in the treatment of chronic autoimmune disease.
As Part 3 highlights, the technological toolkit is expanding faster than clinical registries can keep pace. From the early success of academic autologous CAR-T cells to the rapid advancement of next-generation, multi-targeted TCE platforms engineered to bypass long-lived plasma cell resistance, the ultimate destination remains the same: a multi-year immune reset. As clinical-stage candidates like rese-cel, KYV-101, and an array of novel TCEs push through parallel global trials, the stage is set to extinguish the heat of autoimmune disease for good.
If you missed Part 1 of this three-part Frontiers in Medicine series on CAR-T, check it out here. We discussed the Immunology Civil War, the discovery of T-cells, and their rebirth as medicines. Make sure you check out Part 2 if you haven’t already, where we discuss the evolution of therapeutic modalities beyond autologous CAR-T: allogeneic, in vivo, and T-cell engagers (TCE).
To contact us, please send us an email at biotechreadout@gmail.com
Disclaimers
Investigational Status Disclaimer
The therapeutic candidates discussed in this newsletter are currently in clinical development and have not been approved for commercial sale by the U.S. Food and Drug Administration (FDA), the European Medicines Agency (EMA), or other global regulatory authorities. Their safety and efficacy have not been established. References to pipeline products and ongoing clinical trials involve significant risks and uncertainties. Statements regarding the potential safety, potency, or efficacy of investigational drugs reflect current hypotheses and are not a guarantee of future performance or regulatory clearance. The outcome of clinical trials is inherently unpredictable, and clinical results from earlier stages may not be predictive of results in later, larger-scale trials.
No Medical Advice Disclaimer
This newsletter is for informational and educational purposes only. The content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or another qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read in this publication.
No Patient-Provider Relationship Disclaimer
The information provided in this newsletter is for educational and analytical purposes only. Receipt of this information, or any interaction with this content, does not create a physician-patient, pharmacist-patient, or any other professional-provider relationship between you and the authors or publishers. This newsletter should not be used as a substitute for a personal consultation with a qualified healthcare professional.
Forward-Looking Statements Disclaimer
This newsletter contains “forward-looking statements” regarding future events, including clinical trial timing, regulatory milestones, and projected market performance. These statements are based on current expectations and assumptions that are subject to significant risks and uncertainties. Actual results may differ materially from those expressed or implied. We undertake no obligation to update these statements as a result of new information or future developments.
Third-Party Links & Content Disclaimer
This newsletter contains links to third-party websites, including clinical trial registries and corporate presentations. Biotech Readout does not endorse, guarantee, or assume responsibility for the accuracy or reliability of any information offered by third-party providers.
Errors and Omissions Disclaimer
While we strive for technical accuracy, the information in this newsletter is provided on an “as is” basis with no guarantees of completeness, accuracy, or timeliness. Biotech Readout assumes no liability for any errors or omissions in the content of this publication.
Non-Endorsement Disclaimer
Any reference to specific commercial products, processes, or services by trade name, trademark, or manufacturer does not constitute or imply an endorsement or recommendation by the author. All trademarks are the property of their respective owners.
No Investment Advice Disclaimer
This newsletter is for informational purposes only and does not constitute financial, investment, or legal advice. The author is not a registered investment advisor. You should consult with a professional financial advisor before making any investment decisions. The biotechnology sector is highly volatile; past performance is not indicative of future results.
Conflict of Interest Disclaimer
The author of this newsletter maintains a position of independence. At the time of publication, the author holds no direct financial interest, equity, or options in any of the companies mentioned in this report. No compensation has been received from any third party to feature or analyze specific therapeutic candidates or corporate entities.