
CAR-T, Part 1
Immunology Civil War, the discovery of T-cells, and their rebirth as medicines

Disclaimer: This newsletter is for educational and informational purposes only and does not constitute medical, investment, or financial advice, nor does it establish a provider-patient relationship. Content may include forward-looking statements and discussions of investigational therapeutic candidates that are not FDA/EMA approved; their safety and efficacy remain unestablished and clinical outcomes are unpredictable. While we strive for accuracy, all information is provided as is without guarantees. This newsletter is independent, and the author holds no financial positions in the companies mentioned nor receives third-party compensation for this coverage. Please find a complete version of our disclaimers at the bottom of this article and on our About page.

Introduction
On a wintry Friday evening in Stockholm, Ilya Mechnikov approached the podium at the Royal Academy of Music and delivered his Nobel Lecture before an audience of distinguished scientists and Swedish royalty. It was a speech that would end a decades-long civil war within the field of immunology, and would change the course of history forever.
“There is no need to be a doctor or a scientist to wonder why the human body is capable of resisting so many harmful agents in the course of everyday life. It is often seen that in households where all members are exposed to the same danger [whilst] disease does not strike everyone indifferently. For some individuals who go down at the attack, there are others who have immunity to a greater or lesser extent. […] A battle takes place between the two elements. […] Whenever the organism enjoys immunity, the introduction of infectious microbes is followed by the accumulation of mobile cells, of white corpuscles of the blood in particular which absorb the microbes and destroy them.”
Ilya Mechnikov’s Nobel Lecture (December 11, 1908)
Mechnikov, who was working at the Pasteur Institute in Paris at the time, had discovered that specific cells or, “white corpuscles of the blood” as he called them, moved toward and engulfed foreign particles. Before Mechnikov, inflammation (redness, heat, swelling) was viewed by the medical establishment as a purely destructive process, a symptom of disease. However, Mechnikov flipped this view. He argued that inflammation is a healing reaction. He demonstrated that the swelling and heat were actually signs of the body’s cellular army mobilizing to the site of injury.
Mechnikov saw the immune system as an ancient, inherited defense; a battle that had been raging since the first multicellular organisms appeared. However, the discovery of T-cells decades later revealed a complexity he could only imagine. By merging Mechnikov’s cellular army with the pinpoint targeting of laboratory engineering, we have moved beyond simply observing the battle to actively directing it.
Cells fighting cells. Cells fighting bacteria. Cells fighting cancer.
In Part 1 of this three-part Frontiers in Medicine series on CAR-T, we dive into the Immunology Civil War, discovery of T-cells, and their rebirth as medicines.
Stay tuned for Part 2, where we discuss the evolution of therapies that extend the legacy of CAR-T, and Part 3, where we discuss the re-tooling of CAR-T against autoimmune diseases.
Immunotherapy’s Humble Beginnings
The history of Chimeric Antigen Receptor T-cell therapy (CAR-T) is a long and winding journey from early 19th-century observations to the cutting-edge techniques being developed today. The story begins with in the 1890s with Dr. William B. Coley, a bone surgeon at New York Memorial Hospital (now called Memorial Sloan Kettering Cancer Center).
In 1891, Dr. Coley was deeply affected by a young patient who had died due to bone cancer. While searching through hospital records to look for patients who had better outcomes, he found a case from seven years prior: a patient with an inoperable tumor in the neck had made a full recovery after contracting Erysipelas (a skin infection caused by Streptococcus pyogenes). Dr. Coley tracked the man down and found him healthy and cancer-free. He hypothesized that the severe immune response triggered by the infection had incidentally destroyed the cancer cells. Dr. Coley began experimenting by intentionally infecting cancer patients with live bacteria. While he saw some success, this was dangerous; at least two patients died from the infections. To make the treatment safer and more predictable, he switched to a mixture of heat-killed bacteria consisting of Streptococcus pyogenes (Gram-positive) and Serratia marcescens (Gram-negative). This cocktail became known as Coley’s Toxins. It was designed to provoke a high fever and a massive systemic immune response without causing a full-blown infection.
At the time, Coley didn’t know the molecular biology behind his treatment. Today, we understand that Coley’s Toxins worked through several immunological pathways:
Cytokine Storm: The bacterial components (specifically Lipopolysaccharides or LPS) triggered the release of Tumor Necrosis Factor (TNF), Interleukins, and Interferons.
Dendritic Cell Activation: The toxins acted as adjuvants, activating the innate immune system to recognize tumor antigens that it had previously ignored.
Fever Therapy: The sustained high fever (often reaching 103–105°F) induced by the toxins is thought to have a direct inhibitory effect on some tumor types and further stimulate immune cell activity.
During his career, Coley treated nearly 1,000 patients. Many cases of inoperable sarcomas (bone and soft tissue cancers) went into complete remission. However, the therapy faced significant criticism and was ultimately abandoned by the medical establishment due to several critical flaws:
Inconsistency: Coley frequently changed the formula and dosage, making it difficult for other doctors to replicate his results.
The Rise of Radiation: At the turn of the 20th century, X-rays and radium were discovered. Radiation was more modern, easier to standardize, and yielded more immediate results, leading the medical community to favor it over unpredictable toxins.
The AMA Critique: The American Medical Association was skeptical of the treatment due to its lack of controlled trials, and the therapy was eventually labeled as “unproven”.
While Dr. William Coley was on the right track in identifying a connection between cancer and the immune system, he lacked a understanding of why Coley’s Toxins sometimes worked and how it could be optimized. In the 1970s, Dr. Lloyd Old provided another piece to the puzzle by utilizing a weakened strain of Mycobacterium bovis (the BCG vaccine) to train the immune system to attack non-muscle invasive bladder cancer (NMIBC). The success of the BCG vaccine in bladder cancer was not due to the vaccine attacking the cancer directly. Instead, it functioned as a biological adjuvant. When BCG is instilled directly into the bladder, it infects the urothelial cells and the tumor cells. This infection triggers a massive local release of cytokines (like IL-2, IL-12, and IFN-gamma). These signals act as a beacon, recruiting T-cells, Natural Killer (NK) cells, and macrophages to the bladder wall. The immune system, now highly activated to fight the bacterial infection, recognizes and destroys the tumor cells in the process.
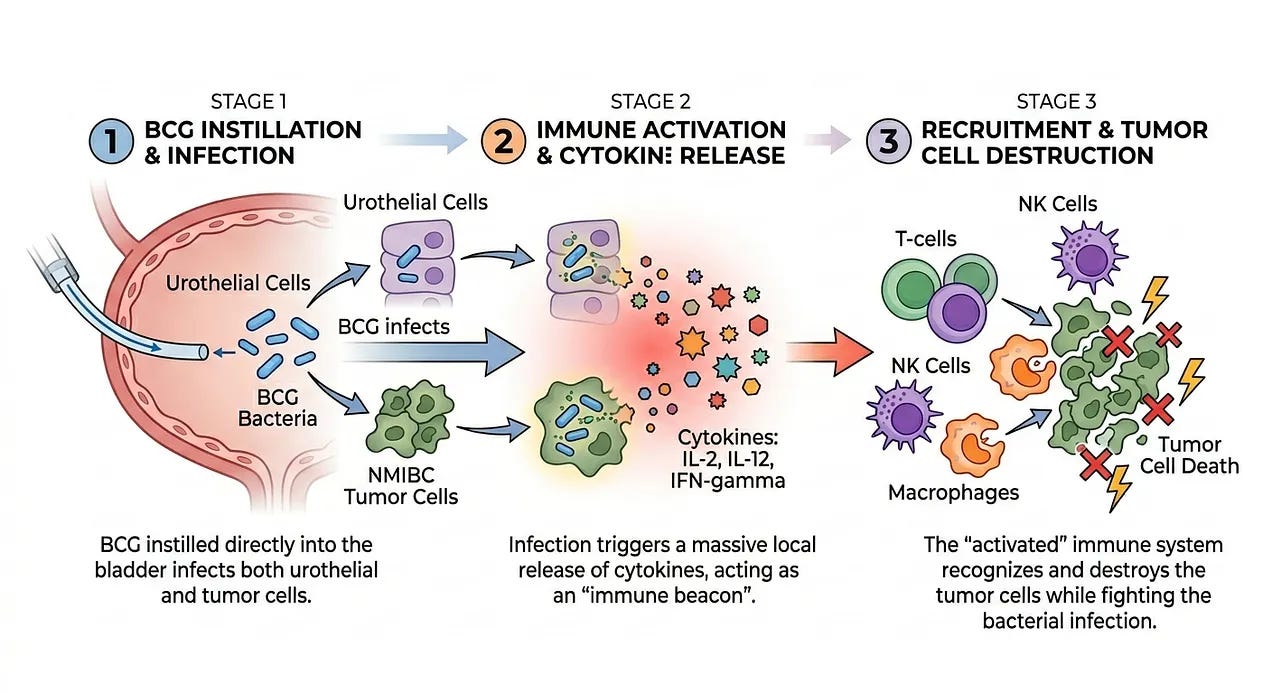
Dr. Old’s work was pivotal for three reasons that paved the way for CAR-T therapies:
Validation of Non-Self Recognition: It proved that if you could make a tumor visible to the immune system, the body had the inherent machinery to eliminate it.
The Birth of “Active” Immunotherapy: Unlike non-living therapies like chemotherapy or later monoclonal antibodies, BCG required the patient’s own T-cells to do the heavy lifting, a fundamental requirement for CAR-T.
FDA Precedent: In 1990, BCG became the first immunotherapy to receive FDA approval for cancer, transforming it from an experimental theory into a standard of care that remains the frontline treatment for non-muscle invasive bladder cancer (NMIBC) today.
Yet, the discovery of Coley’s Toxins and the BCG vaccine begs the question: what is the mechanism of immunity? This very question set off a decades-long civil war, a bitter rivalry between two schools of thought in the field of immunology.
Immunology Civil War: Cellularists versus Humoralists
The “Immunology Civil War” was a decades-long intellectual battle at the end of the 19th century that fractured the scientific world into two hostile camps. It was a disagreement that transcended science alone and spilled over into clashing cultures, nationalities, and philosophies about how life defends itself. The conflict pitted two camps against each other and was sharpened by the borders of 19th-century Europe:
Humoralists: They were led by giants like 1905 Nobel Laureate Robert Koch, 1901 Nobel Laureate Emil von Behring, and soon-to-be 1908 Nobel Laureate Paul Ehrlich. Humoralists belonged to the German school of thought, which was rooted in chemistry. They saw the body as a sophisticated laboratory. To them, immunity was a series of precise, predictable chemical reactions: neutralization, precipitation, and side-chain interactions.
Cellularists: They were led by the Russian zoologist and Paul Ehrlich’s soon-to-be 1908 Nobel co-Laureate Ilya Mechnikov. He belonged to the French school of thought, which was rooted in biology and followed in the footsteps of Louis Pasteur, who first published on the Germ Theory of Disease in 1857 and whose passing in 1895 predated the establishment of the Nobel Prize in 1901 by a mere six years. Cellularists saw the body as a living, breathing ecosystem. To them, immunity was an active, Darwinian struggle where cells decided to attack invaders.
In the 1890s, the Humoralists held the political upper hand. Emil von Behring had discovered antitoxins for diphtheria and tetanus. They showed that the liquid part of the blood (the serum) could neutralize toxins even without any cells present. The Germans ridiculed Mechnikov’s “wandering cells.” They argued that “phagocytes” (immune cells that engulfed and destroyed foreign particles, bacteria, and dead/dying cells) were merely scavengers that cleaned up the mess after the chemical serum had already killed the bacteria. In some cases, they believed white blood cells actually spread disease by eating bacteria and carrying them alive to other parts of the body.
Mechnikov, working out of the Pasteur Institute in Paris, countered with a radical idea: the cell was the instigator. He spent years performing meticulous microscopy to show that white blood cells actively hunted bacteria (chemotaxis) rather than simply stumbling upon them. The “humors” (serum) only seemed effective because the cells had produced the protective substances in the first place or were aided by them.

The war finally began to de-escalate in the early 1900s through the work of British scientist Almroth Wright. He discovered substances he called opsonins (from the Greek “to prepare food for”). Wright demonstrated that the serum (Humoral) contained so-called “opsonins” that coated the bacteria. This coating made the bacteria “tasty” and easier for the white blood cells (Cellular) to grab and eat. This unified theory suggested that that both sides were right.
This Immunology Civil War was tense but essential because it forced both sides to engage in an arms race that clarified each theory independently, until they were ultimately bought together into one harmonious model.
For the Cellularists, it led to the discovery of the Innate Immune System, the rapid, non-specific response we are born with. Dendritic Cells or Macrophages use general sensors to detect detect non-self invaders, but they are blind to specific invaders. They eat a non-self invader and, importantly, show pieces of it (“antigens”) to the rest of the army.
For the Humoralists, it led to the discovery of the Adaptive Immune System, the slower but more specific response that identifies invaders and remembers them. Triggered by the innate cells, B-cells/plasma cells churn out antibodies and T-cells that destroy the invaders.
The 1908 Nobel Prize was a deliberate “peace prize,” awarded jointly to Mechnikov (Cells) and Ehrlich (Humors), formally acknowledging that the human body requires both its chemical weapons and its cellular soldiers to survive. By awarding the prize to both Ilya Mechnikov and Paul Ehrlich, the Nobel Committee was forcing a reconciliation between two seemingly irreconcilable worldviews. It served as a formal declaration that the immune system was integrated and transitioned the field from “either/or” to “both/and.” This détente taught scientists valuable lessons that led to the intellectual maturity of immunology:
Complexity is the rule: Evolution rarely relies on a single defense mechanism.
Redundancy is a feature: The body has chemical weapons and cellular soldiers because if one fails, the other can often compensate.
Cross-Disciplinary Discovery: The most powerful breakthroughs happen at the intersection of different fields (in this case, Zoology and Organic Chemistry). Immunology transitioned from a field of competing silos to one of integrated systems.
Uncovering Immunity’s Cellular & Molecular Roots
As we have seen, until the mid-20th century, scientists believed the immune system was a single, monolithic entity. However, the discovery that it was actually composed of specialized cell types changed medicine forever. But how did we identify those cell types, and how do they differentiate between friend and foe? It starts with a forgotten organ.
For decades, the thymus (a small organ in the upper chest) was considered a vestigial organ, essentially a useless evolutionary leftover. The breakthrough came in 1961 with Dr. Jacques Miller, a French-Australian research scientist whose pivotal work occurred early in his career as a Ph.D. student at the University of London. He was studying leukemia in mice and began removing the thymus of newborn mice in a procedure known as a neonatal thymectomy. He observed something startling: the mice without a thymus were unable to reject foreign skin grafts, had almost no lymphocytes (white blood cells) in certain areas of their lymph nodes, and died prematurely from common infections. This provided evidence that the thymus was the “schoolhouse” where a specific type of immune cell learned how to function. As an aside, Jacques Miller is often cited as the only person still living who discovered the function of a major human organ. It is widely considered one of the greatest snubs in Nobel Prize history that he hasn’t received the award for medicine yet, though he did receive the Lasker Award in 2019.
Around the same time, Dr. Max Cooper and Dr. Robert Good were studying the Bursa of Fabricius in chickens (an organ humans don’t have, but which serves as the school for B-cells). They realized that the immune system had two distinct branches:
B-cells (Bursa-derived): These cells produced antibodies to attack invaders in the blood (humoral immunity).
T-cells (Thymus-derived): These cells did not make antibodies. Instead, they attacked infected or non-healthy cells directly (cell-mediated immunity).
While the work of Drs. Jacques Miller and Max Cooper in the 1960s successfully identified the T-cell as the footsoldier of the immune system, it simultaneously birthed one of the greatest mysteries in modern biology: how did the T-cell see its target? Unlike B-cells, which secrete antibodies that circulate like free-floating heat-seeking missiles, T-cells require physical, cell-to-cell contact to strike. Scientists hypothesized that this implied a physical receptor on their surface. Yet for two decades, this receptor remained unknown. The hunt moved from the gross anatomy of the thymus to the microscopic realm of the cell membrane and the hypothesized T-cell Receptor (TCR).
Before scientists had identified the receptor, they witnessed its peculiar behavior. In 1974, Peter Doherty and Rolf Zinkernagel discovered MHC Restriction. They found that T-cells were “double-locked”: they didn’t just recognize a virus; they recognized a virus only when it was presented by the body’s own self-identity markers (MHC). This was a massive hurdle for early researchers because it suggested the TCR wasn’t a simple key and lock like an antibody, but a complex sensor capable of a “dual handshake”. This work was so foundational it eventually earned the Nobel Prize in 1996.
By the early 1980s, the race became a sprint to physically isolate the receptor protein. Since the human genome hadn’t yet been sequenced, researchers used monoclonal antibodies as hooks to see if they could grab a protein on the T-cell surface that differed from one cell to the next. The breakthrough came in 1982, when Dr. James Allison (who later won the Nobel Prize for discovering immune checkpoint inhibitors) identified a protein that was “clonotypic”, meaning it was unique to a specific line of T-cells. Shortly after, teams led by Ellis Reinherz, and the duo of Philippa Marrack and John Kappler, confirmed the TCR was a heterodimer composed of two chains (alpha and beta) linked together.
The climax of this history occurred in 1984, when the genetic blueprints for the TCR were finally cloned. Two labs working independently on opposite sides of the world crossed the finish line almost simultaneously: Dr. Mark Davis (Stanford) and Dr. Tak Wah Mak (University of Toronto). They used a brilliant technique called subtractive hybridization to compare the mRNA of T-cells to that of B-cells. Since the cells are nearly identical except for their receptors, they subtracted the common genes. What remained was the code for the TCR.
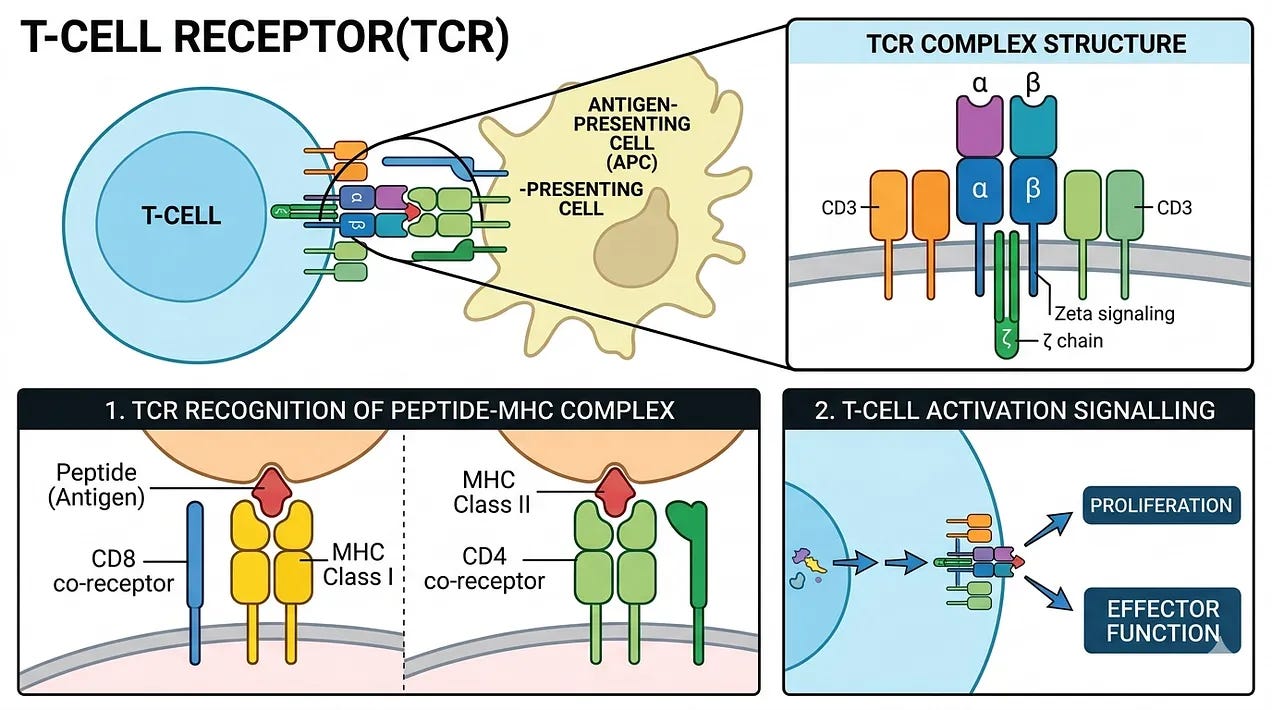
So, what is our most up-to-date understanding of how T-cells find and eliminate infected cells or tumor cells? T-cells circulate through the body, constantly feeling the surface of other cells. They aren’t looking for the virus/tumor itself, but for a specific signal called the MHC-peptide complex. Every cell in your body regularly chops up its internal proteins into tiny pieces (peptides) and holds them out on a tray called the MHC (Major Histocompatibility Complex). If the cell is healthy, the T-cell’s TCR recognizes the peptide as “self” and moves on. When a cell is infected or cancerous, it inadvertently puts a piece of the viral or mutated protein on that MHC tray. If a T-cell has a TCR that perfectly fits that specific viral or mutated peptide, it latches on. This is the MHC Restriction mentioned earlier, the TCR must recognize both the foreign peptide and the body’s own MHC tray simultaneously. This ensures the T-cell only attacks infected body cells, not free-floating viruses (which are the B-cell’s job). Once the TCR is locked in, the T-cell undergoes a physical transformation to become a Cytotoxic T-Lymphocyte (CTL). The T-cell pulls the infected cell close, forming an immunological synapse, a tight seal between the two. The T-cell sprays toxic proteins into the gap, primarily perforins (pokes holes in the infected cell’s membrane) and granzymes (enters through perforin holes and triggers programmed cell death or apoptosis). The infected cell implodes, preventing the virus or cancer from replicating further. The T-cell then detaches, completely unharmed, and moves on to find the next target.
Programming T-cells to Hunt Cancer
The discovery of the T-cell and the subsequent mapping of its receptor (TCR) unveiled a biological assassin designed to eliminate threats while sparing the host. However, cancers deploy strategies to hide from, shield themselves from, and sabotage T-cells that are actively hunting them:
Hiding (avoiding detection): T-cells can only see cancer if the tumor displays its mutated proteins on an MHC tray. Many tumors evolve to stop producing MHC molecules entirely or mutate the proteins (B2M) that stabilize them. Without the tray, the T-cell simply walks past the tumor, unaware it is dangerous. Tumors may also stop expressing the specific mutated proteins (antigens) that the immune system was originally trained to recognize, a phenomenon known as antigen escape.
Shielding (protect itself from harm): Healthy cells have “off switches” called immune checkpoints (like PD-L1 or CD80/CD86) that are displayed on the cell surface and prevent T-cells from attacking. This is useful for preventing the immune system from damaging healthy organs. However, cancer cells frequently over-produce immune checkpoints, forcing hostile T-cells into a state of exhaustion or paralysis. Cancer often utilizes another shielding tactic: the recruitment of immunosuppressive cells that similarly serve to wind down an immune response. These cells include regulatory T-cells (Tregs), which are attracted to tumor-derived TGF-beta and IL-10, and myeloid-derived suppressor cells (MDSCs), which neutralize the toxic proteins T-cells use to kill their target cells (perforin/granzymes).
Sabotaging (actively shutting down the immune system): The area surrounding a tumor, the Tumor Microenvironment (TME), is often transformed into a hostile no-go zone for immune cells. Tumors can recruit cells called fibroblasts to create a dense wall of collagen that physically prevents T-cells from entering the tumor core. Tumors consume vast amounts of glucose and pump out lactic acid, creating an acidic, low-oxygen environment. This toxic soup inhibits T-cell metabolism, making them too weak to mount an attack.
By the late 1980s, the realization was clear: to defeat an opponent as adaptive as cancer, the T-cell’s natural hardware (the MHC-restricted TCR) required a fundamental update. This realization birthed a new era of synthetic immunology. Scientists sought to bypass the tumor’s cloaking devices by creating a hybrid molecule that never existed in nature. By fusing the relentless killing power of the T-cell with the pinpoint targeting precision of a laboratory-engineered antibody, researchers created the first living drug. This was no longer a therapy that merely nudged the immune system to target cancer; it was a genetically fortified army designed to execute its mission with mechanical precision.
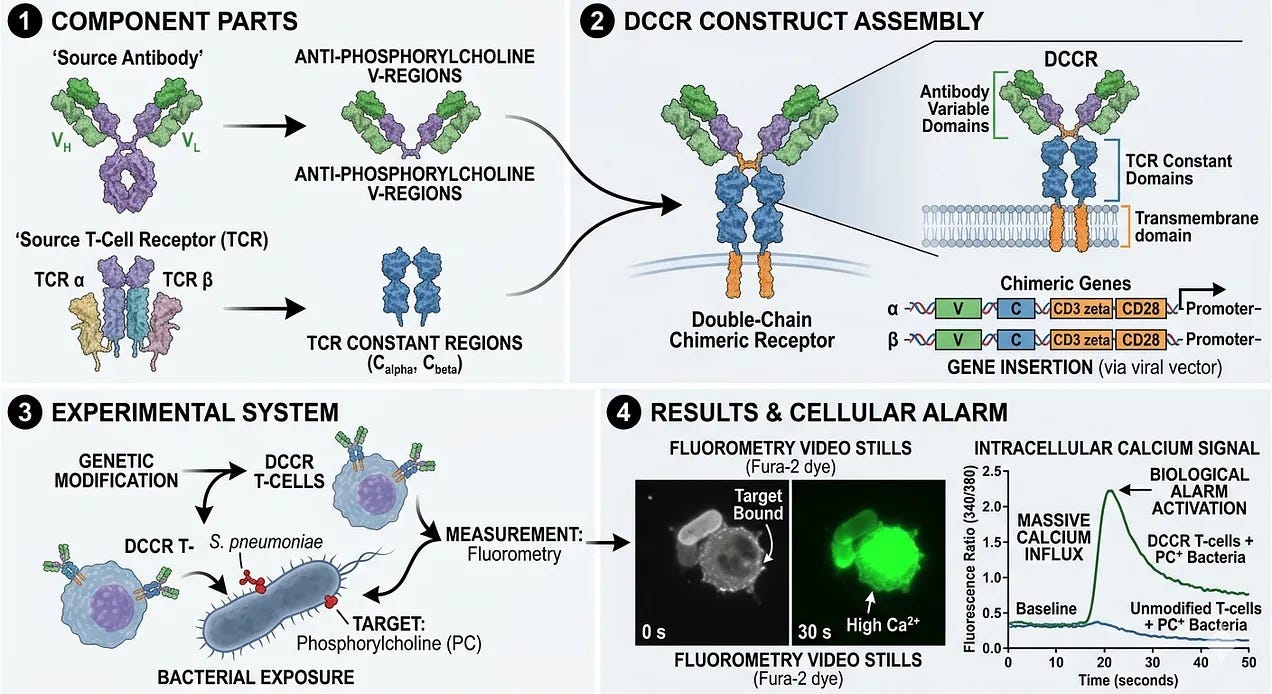
In 1987, Dr. Yoshikazu Kurosawa and his team at the Institute for Comprehensive Medical Science in Japan achieved the world’s first successful fusion of antibody and T-cell receptor components, although it targeted bacteria instead of cancer. Dr. Kurosawa’s team was the first to address the “MHC problem” by asking if a T-cell could be forced to use an antibody’s search function. Unlike modern CAR-Ts, which use a single, simplified chain (scFv), Dr. Kurosawa’s team created a double-chain chimeric receptor (DCCR). They took the Variable (V) regions of an antibody (which recognize specific targets) and fused them to the Constant (C) regions of a T-cell receptor’s Alpha and Beta chains. They chose a target called phosphorylcholine, a molecule found on the surface of certain bacteria (S. pneumoniae). They inserted these chimeric genes into a line of T-cells and exposed them to the bacteria. Using a technique called fluorometry, they watched the cells’ internal chemistry. When the antibody grabbed the bacteria, the T-cells responded with a massive calcium influx, the biological alarm that tells a T-cell it has found a target. This was the first demonstration that a T-cell could be programmed to recognize a target of interest.
Separately, Dr. Zelig Eshhar was working at the Weizmann Institute of Science in Israel. He was frustrated by the fact that T-cells were fussy, they could only see cancer if it was presented on an MHC tray. He knew that antibodies, on the other hand, were expert hunters that could find targets anywhere. To solve this problem, he created a so-called T-Body by fusing the “searching” part of an antibody (known as the single-chain variable fragment or scFv) and the “killing” part of a T-cell (the cytoplasmic domain of the CD3ζ chain that triggers a signaling cascade within a T-cell leading to the release of cell-killing perforin and granzymes). In 1989, Dr. Eshhar published a landmark paper showing that these T-bodies could kill cancer cells in a petri dish. He had effectively bypassed the cloak of invisibility that tumors used to hide from the natural immune system.
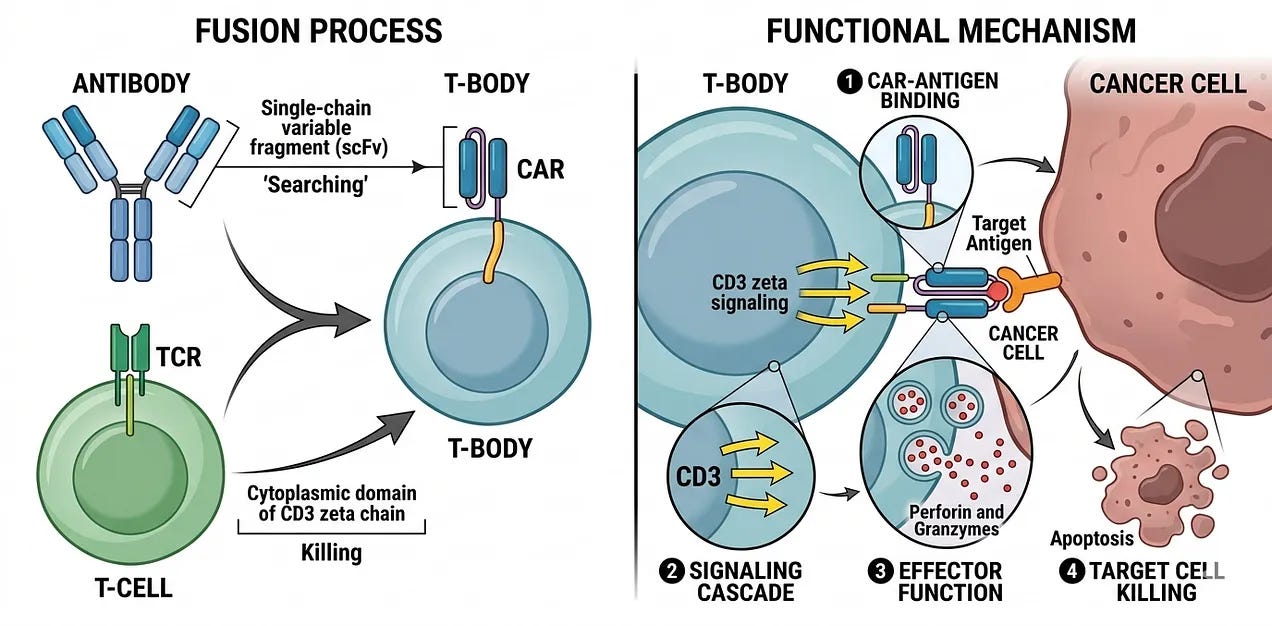
Despite the success in the lab, this 1st generation of CAR-T was a clinical failure when tested in patients throughout the 1990s and early 2000s. Why? Eshhar’s CAR-Ts provided a signal to attack target cells, but they would immediately exhaust themselves and die within hours or days. They couldn’t multiply, and they couldn’t survive long enough to fully eliminate tumors. To get around this, doctors had to give patients extra doses of IL-2, but they caused massive, toxic side effects for the patients, making the treatment both ineffective and dangerous. To kill a large tumor, a few million CAR-T cells would have to turn into billions of CAR-T cells inside the patient.
As a result of these failures, many in the scientific community wrote off CAR-T as a dead end for nearly a decade. Dr. Eshhar himself faced skepticism and struggled to find funding, at one point even pitching an antibody-based opium sensor to a Swedish company just to keep his lab’s lights on. However, this early experiment was essential. It proved that programmed recognition and killing of cancer cells was possible.
It wasn’t until the early 2000s that Dr. Michel Sadelain at Memorial Sloan Kettering and Dr. Carl June at the University of Pennsylvania added the proliferation signals to create 2nd generation CAR-Ts. The breakthrough was the realization that researchers could engineer these signals directly into the synthetic receptor by adding a costimulatory domain between the scFv (targeting) and the CD3ζ chain (killing trigger) domains. In 2002, Dr. Sadelain’s team was the first to demonstrate that adding a CD28 costimulatory domain allowed T-cells to multiply and survive in the presence of cancer. CD28-based CARs are characterized by rapid, powerful metabolic activity. They rely heavily on glycolysis (burning sugar quickly) to mount a massive, immediate attack on the tumor. This design became the foundation for Yescarta (axicabtagene ciloleucel), the first CAR-T approved for lymphoma.
A few years later, Dr. Carl June’s team utilized a different domain called 4-1BB (also known as CD137). 4-1BB is a member of the TNF-receptor family. It sends a different survival signal that promotes oxidative phosphorylation, a slower and more efficient mechanism for burning energy. 4-1BB CARs don’t strike as hard or as fast as CD28 cells, but they have much higher persistence. In some of June’s early patients, these cells were still circulating and patrolling the body ten years after the initial treatment. This design became the foundation for Kymriah (tisagenlecleucel), the first-ever FDA-approved CAR-T therapy (2017).
Cell-ebrating Clinical Success
The 2010 Landmark Trial (published in the New England Journal of Medicine in 2011) was the watershed moment that shifted CAR-T therapy from a theoretical promise to a clinical breakthrough. While the trial was small, enrolling only three patients with advanced Chronic Lymphocytic Leukemia (CLL), the results were so explosive that they fundamentally changed the trajectory of oncology.
By 2010, the three patients in the trial had exhausted every available treatment, including multiple rounds of chemotherapy and monoclonal antibodies. They had several pounds of tumor mass in their bodies and were essentially in end-of-life care. Dr. Carl June’s team utilized their 2nd generation CAR-T design, which featured the 4-1BB costimulatory domain. This was the critical difference; previous trials by other groups using 1st generation CARs had failed to show any significant cell persistence or clinical benefit. After the patients were infused with their own engineered T-cells, the researchers observed a phenomena never before seen in human medicine:
Massive T-cell Expansion: In the patient known as “Patient 1” (Bill Ludwig), the CAR-T cells multiplied by a factor of 1,000 to 10,000 times inside his body.
Serial Killing Effect: Calculations suggested that each single CAR-T cell successfully destroyed approximately 1,000 to 1,500 leukemia cells.
Systemic Eradication: Within weeks, the pounds of tumor mass residing in the blood, bone marrow, and lymph nodes became undetectable.
When the results were published in August 2011, the headlines were staggering. The paper reported Complete Remissions in two of the three patients. Following this trial, Novartis partnered with UPenn in a landmark deal, signaling the first major Big Pharma entry into the space. The 4-1BB domain worked as intended. The CAR-T cells didn’t just kill the cancer and disappear; they remained in the patients’ blood as sentinel cells.
The trial also provided the first clinical description of Cytokine Release Syndrome (CRS). As the CAR-T cells engaged in a massive war with the leukemia, the patients developed high fevers, dangerously low blood pressure, and organ stress. This was a terrifying moment for the researchers, but they soon realized it was paradoxically a biomarker of success: the sicker the patient got, the more effectively the living drug was working.
In a follow-up more than a decade later in 2022, these same patients were still cancer-free, and the original CAR-T cells were still detectable in their blood, proving that a single infusion could potentially provide a lifetime of protection. The study focused on Doug Olson and Bill Ludwig, the two survivors from the 2010 trial. At the 10-year mark, both were still in complete remission (Bill Ludwig remained cancer-free but unfortunately passed away in January 2021 due to complications from COVID-19). Researchers found that the CAR-T cells had evolved inside the patients’ bodies. While killer CD8+ T-cells did the initial work of destroying the tumor in 2010, the long-term patrolling was carried out by a highly activated population of CD4+ CAR T-cells. Following this paper, Dr. Carl June made headlines by stating, “We can now conclude that CAR-T cells can actually cure patients with leukemia,” a rare and bold claim in oncology.
The story of Emily Whitehead is another critical milestone in the development of CAR-T therapy. If the 2011 NEJM paper demonstrated that CAR-T could work in adults, Emily demonstrated that it could work in children, and in doing so, she inadvertently taught the medical world how to manage the therapy’s most dangerous side effect: Cytokine Release Syndrome (CRS).
Emily was diagnosed with acute lymphoblastic leukemia (ALL) in 2010 at age five. Despite 16 months of aggressive chemotherapy, her cancer relapsed twice. By early 2012, her doctors told her parents, Tom and Kari Whitehead, that she had run out of options and should enter hospice. Refusing to give up, they enrolled her in a Phase 1 clinical trial at Children’s Hospital of Philadelphia (CHOP) led by Dr. Stephan Grupp. On April 17, 2012, Emily became the first pediatric patient in the world to receive CAR-T cells (the same 4-1BB construct developed by Carl June’s team).
A few days after the infusion, Emily became critically ill. This was the first time pediatricians had witnessed a full-scale Cytokine Release Syndrome (CRS). She experienced 105°F fevers, a massive drop in blood pressure, and respiratory failure. She was placed on a ventilator in the PICU and given a 1-in-1,000 chance of surviving the night. Dr. Grupp’s team ran an emergency blood test and found that her levels of Interleukin-6 (IL-6), a signaling protein for inflammation, were 1,000 times higher than normal. In one of the most famous happy accidents in medical history, Dr. Carl June’s own daughter had juvenile arthritis and took a drug called Actemra, which happens to block IL-6. Actemra had never been used for cancer-related inflammation, but with Emily near death, the team administered it as a desperate measure. Within hours, her fevers vanished. She woke up on May 2, 2012, her 7th birthday. If Emily had died, it is widely believed the FDA would have shut down the CAR-T program. Instead, her survival solidified Actemra as the gold standard of managing CRS, though the condition remains a significant and potentially life-threatening risk of CAR-T therapy.
Three weeks after Emily woke up, a bone marrow biopsy showed no signs of cancer. Emily famously stood by her father’s side when he testified before the FDA in a 2017 hearing, leading to the unanimous approval of Kymriah, the first approved CAR-T therapy. Like the adult patients in the 2022 Nature study, Emily still has patrolling CAR-T cells in her blood today. They have functioned as a permanent, living immune system against her leukemia for over a decade.
As of April 2026, the FDA has approved several CAR-T therapies that have transformed the treatment landscape for hematologic malignancies. These therapies are generally categorized by their target antigen: CD19 (for B-cell leukemias and lymphomas) and BCMA (for multiple myeloma).
CD19 is a protein found on the surface of nearly all B-cells, both healthy and cancerous. This makes it an ideal target for B-cell related cancers. These are the FDA-approved CAR-T therapies targeting CD19:
Kymriah (Tisagenlecleucel): The “pioneer.” Approved in 2017, it uses a 4-1BB costimulatory domain (the design from Carl June’s team). It is primarily used for pediatric and young adult ALL and certain large B-cell lymphomas.
Yescarta (Axicabtagene ciloleucel): Also approved in 2017, it uses a CD28 costimulatory domain (the Sadelain design). It is a standard of care for aggressive Large B-cell Lymphoma (LBCL) and Follicular Lymphoma (FL).
Tecartus (Brexucabtagene autoleucel): Developed specifically for Mantle Cell Lymphoma (MCL) and adult ALL. It involves a specialized manufacturing process that removes circulating tumor cells to prevent them from interfering with the T-cell engineering.
Breyanzi (Lisocabtagene maraleucel): Notable for its defined composition. Unlike other CAR-Ts where the ratio of CD4+ to CD8+ cells is random, Breyanzi is administered in a specific 1:1 ratio. This is intended to make the side-effect profile (like CRS) more predictable and manageable.
For decades, Multiple Myeloma was considered incurable. The discovery of B-cell Maturation Antigen (BCMA) as a target allowed CAR-T to enter this space. These are the FDA-approved CAR-T therapies targeting BCMA:
Abecma (Idecabtagene vicleucel): The first BCMA-directed CAR-T approved (2021). It is used for patients with relapsed or refractory multiple myeloma who have already tried four or more lines of therapy.
Carvykti (Ciltacabtagene autoleucel): A highly potent dual-target therapy. It features two different BCMA-binding domains that attach to the cancer cell in two different spots. Carvykti has shown some of the highest response rates in clinical history for myeloma, with a recent trend moving it into earlier lines of treatment (meaning patients get it sooner in their journey, rather than as a last resort).
Scientists continue to innovate in the space with novel targets for hematological malignancies (CD79a, PRC5D, CMA, CD33) and solid tumors (CLDN6, CLDN18.1, MUC16, GPC3, PSMA) that are currently being tested in clinical trials.
CAR-T’s Challenge #1: Access Gap
Emily Whitehead’s father often speaks about the lottery they won by being near Children’s Hospital of Philadelphia (CHOP) and having access to the Kymriah clinical trial. Therein lies the biggest challenge for CAR-T therapy. While CAR-T is a medical breakthrough, it is currently a logistical nightmare. The biotech industry refers to this as the Access Gap: a chasm between the number of patients who need the therapy and the number of doses that can actually be manufactured.
At the heart of this problem is the autologous nature of current therapies: every single dose is a bespoke, one-off product created from the patient’s own living cells. The most critical metric in the CAR-T industry is Vein-to-Vein (V2V) time. This is the total number of days between the moment a patient’s cells are collected and the moment they are infused back into the patient as a living drug. The typical vein-to-vein time averages 21 to 35 days and involves:
Leukapheresis: The patient travels to a specialized center where their blood is filtered to collect T-cells.
Cold Chain Logistics: The cells are cryopreserved and shipped via specialized medical couriers to a centralized manufacturing facility (often across the country).
Genetic Re-coding: In a clean Room (ISO 5 environment), technicians use viral vectors to insert the CAR gene into the T-cells.
Expansion: The cells are grown in bioreactors until they reach a therapeutic dose (often hundreds of millions of cells).
Quality Control (QC): The final product undergoes rigorous testing for purity, potency, and sterility.
Return Flight: The cells are frozen again, shipped back, and thawed at the patient’s bedside for infusion.
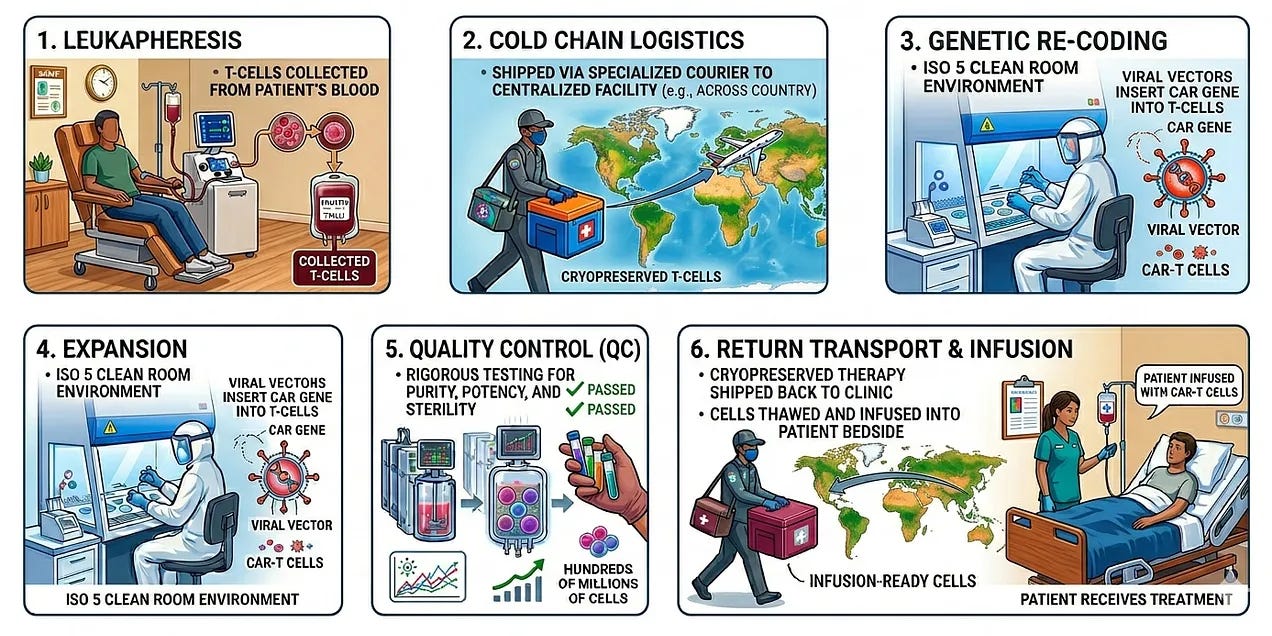
The “vein-to-vein” process is fraught with several points of failure:
Manufacturing Constraints: High-tech manufacturing facilities have limited capacity. A patient might be ready for treatment but, if there is no slot available at the factory, they must wait, often while their cancer continues to progress.
Manufacturing Failure: Since the starting material is a sick patient’s T-cells (which have often been decimated by years of chemotherapy), the cells sometimes fail to grow in the lab. This results in a manufacturing failure, meaning the patient loses weeks of time and may no longer be healthy enough for a second attempt.
The Cold Chain: Maintaining a chain of custody at temperatures below -150°C is incredibly expensive and risky. A single power failure or shipping delay can destroy a half-million-dollar treatment.
Bridging Chemotherapy: Since the process takes a month, many patients require bridging chemotherapy just to keep them alive until their CAR-T cells are ready. This adds further toxicity and cost to the treatment.
Due to the complexity of leukapheresis and CRS management, CAR-T is largely restricted to elite academic medical centers. Patients in rural areas or developing nations are effectively locked out of the therapy. The labor-intensive, one-patient-one-batch model is the primary driver of the astronomical cost of these therapies, which can exceed $400,000. Despite the clinical triumphs of Drs. June and Sadelain, the ‘vein-to-vein’ reality of autologous CAR-T remains present significant challenges to global health equity for the average patient. Ruxandra Teslo wrote an excellent piece on this in the The Clinical Trials Abundance blog, describing how the logistical challenges of manufacturing autologous CAR-T therapy almost derailed J&J/Legend Biotech’s launch of Carvykti.

CAR-T’s Challenge #2: Lymphodepleting Chemotherapy
The next biggest challenge for CAR-T therapy after the access gap is the requirement for lymphodepleting chemotherapy. If you infuse CAR-T cells into a normal immune system, the new cells often fail to expand because they are competing with the existing resident cells. Conditioning serves three primary functions:
Creating a Niche: It physically eliminates existing lymphocytes to make room for the CAR-T cells to multiply.
Cytokine Sink Elimination: It removes cells that soak up vital growth factors like IL-7 and IL-15. By removing the competition, these homeostatic cytokines surge, providing a nutrient-rich environment for the CAR-T cells to feast on.
Suppression of Suppressors: It temporarily wipes out Regulatory T-cells (Tregs) and Myeloid-Derived Suppressor Cells (MDSCs) that the tumor has recruited to protect itself.
While various protocols exist, the gold standard for CAR-T conditioning is the combination of Fludarabine and Cyclophosphamide (often abbreviated as Flu/Cy). Flu/Cy is typically administered over three consecutive days, ending 2 to 7 days before the CAR-T infusion. Unlike myeloablative chemotherapy used for bone marrow transplants, which completely wipes out the bone marrow, lymphodepletion is generally non-myeloablative. It is designed to be intense enough to clear the T-cells but gentle enough that the patient’s blood counts can eventually recover.
However, lymphodepleting chemotherapy adds a significant layer of treatment burden to an already frail patient:
Cytopenias: Patients often experience prolonged periods of low white blood cell counts (neutropenia), leaving them extremely vulnerable to life-threatening infections during the window when the CAR-T cells are starting their work.
Gut Toxicity: Chemotherapy can damage the lining of the gut, which some researchers believe may actually contribute to the severity of Cytokine Release Syndrome (CRS) by allowing bacterial products to leak into the bloodstream.
The convergence of these two challenges, the grueling logistics of the vein-to-vein cycle and the physiological toll of lymphodepleting chemotherapy, creates a formidable ceiling for the current generation of CAR-T therapies. While autologous manufacturing has undoubtedly turned the tide for thousands of patients, it remains a bespoke, resource-intensive model that tethers a revolutionary medicine to high-cost academic centers and toxic pre-conditioning regimens. The future of the field, therefore, hinges on a fundamental decoupling of the therapy from the lab.
Conclusion
The journey from Ilya Mechnikov’s observation of cellular immunity to the precise genetic re-coding of a patient’s own T-cells represents a century of medical progress and a fundamental shift in our relationship with biology. We have moved from being passive observers of the “Immunology Civil War” to the architects of its most advanced weaponry.
However, as we have seen, the intellectual maturity of the field has brought us to a new kind of crossroads. The clinical triumphs of pioneers like Carl June and Michel Sadelain have proved that we can achieve remissions that last years, but the logistical hurdles of the access gap and the physiological toll of lymphodepletion remind us that our work is unfinished. We have created a compelling treatment, but it is currently a bespoke treatment, tethered to elite labs and high-cost academic centers. The next era of CAR-T will not be defined by whether we can kill cancer, but by whether we can decouple the therapy from the laboratory.
If the 20th century was about understanding the “battle” Mechnikov described, the 21st century is about democratizing the victory. Check out Part 2, where we discuss the evolution of therapies that extend the legacy of CAR-T, and Part 3, where we discuss the re-tooling of CAR-T against autoimmune diseases.


{kind=link}
To contact us, please send us an email at biotechreadout@gmail.com
Disclaimers
Investigational Status Disclaimer
The therapeutic candidates discussed in this newsletter are currently in clinical development and have not been approved for commercial sale by the U.S. Food and Drug Administration (FDA), the European Medicines Agency (EMA), or other global regulatory authorities. Their safety and efficacy have not been established. References to pipeline products and ongoing clinical trials involve significant risks and uncertainties. Statements regarding the potential safety, potency, or efficacy of investigational drugs reflect current hypotheses and are not a guarantee of future performance or regulatory clearance. The outcome of clinical trials is inherently unpredictable, and clinical results from earlier stages may not be predictive of results in later, larger-scale trials.
No Medical Advice Disclaimer
This newsletter is for informational and educational purposes only. The content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or another qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read in this publication.
No Patient-Provider Relationship Disclaimer
The information provided in this newsletter is for educational and analytical purposes only. Receipt of this information, or any interaction with this content, does not create a physician-patient, pharmacist-patient, or any other professional-provider relationship between you and the authors or publishers. This newsletter should not be used as a substitute for a personal consultation with a qualified healthcare professional.
Forward-Looking Statements Disclaimer
This newsletter contains “forward-looking statements” regarding future events, including clinical trial timing, regulatory milestones, and projected market performance. These statements are based on current expectations and assumptions that are subject to significant risks and uncertainties. Actual results may differ materially from those expressed or implied. We undertake no obligation to update these statements as a result of new information or future developments.
Third-Party Links & Content Disclaimer
This newsletter contains links to third-party websites, including clinical trial registries and corporate presentations. Biotech Readout does not endorse, guarantee, or assume responsibility for the accuracy or reliability of any information offered by third-party providers.
Errors and Omissions Disclaimer
While we strive for technical accuracy, the information in this newsletter is provided on an “as is” basis with no guarantees of completeness, accuracy, or timeliness. Biotech Readout assumes no liability for any errors or omissions in the content of this publication.
Non-Endorsement Disclaimer
Any reference to specific commercial products, processes, or services by trade name, trademark, or manufacturer does not constitute or imply an endorsement or recommendation by the author. All trademarks are the property of their respective owners.
No Investment Advice Disclaimer
This newsletter is for informational purposes only and does not constitute financial, investment, or legal advice. The author is not a registered investment advisor. You should consult with a professional financial advisor before making any investment decisions. The biotechnology sector is highly volatile; past performance is not indicative of future results.
Conflict of Interest Disclaimer
The author of this newsletter maintains a position of independence. At the time of publication, the author holds no direct financial interest, equity, or options in any of the companies mentioned in this report. No compensation has been received from any third party to feature or analyze specific therapeutic candidates or corporate entities.