
EULAR 2026
Reviewing innovation from Europe's largest immunology conference

Disclaimer: This newsletter is for educational and informational purposes only and does not constitute medical, investment, or financial advice, nor does it establish a provider-patient relationship. Content may include forward-looking statements and discussions of investigational therapeutic candidates that are not FDA/EMA approved; their safety and efficacy remain unestablished and clinical outcomes are unpredictable. While we strive for accuracy, all information is provided as is without guarantees. This newsletter is independent, and the author holds no financial positions in the companies mentioned nor receives third-party compensation for this coverage. Please find a complete version of our disclaimers at the bottom of this article and on our About page.

Press ⬇️ to go down to a section and ⬆️ to go back up to the Table of Contents
Table of Contents
The European Alliance of Associations for Rheumatology (EULAR) 2026 Congress in London just wrapped up (held from June 3–6, 2026), delivering significant Phase 3 readouts, head-to-head data, and updates across key immunology portfolios. It stands alongside the American College of Rheumatology (ACR) convergence as one of the two premier global medical conferences dedicated to breakthroughs in rheumatic and musculoskeletal diseases (RMDs).
Here, we review the following hand-picked clinical data presented at EULAR 2026:
Potentially best-in-disease UCB - Bimzelx (anti-IL-17A/F mAb) versus Skyrizi (anti-IL23 mAb) / Phase 3b (PsA) ⬇️
Potentially first-in-class Biogen - Dapirolizumab (anti-CD40L mAb) / Phase 3 (SLE) ⬇️
Potentially first-in-disease Novel mechanism Zenas BioPharma - Obexelimab (CD19 x FcγRIIb mAb) / Phase 3 (IgG4-RD) ⬇️
Potentially first-in-disease Novartis - Ianalumab (anti-BAFF mAb) / Phase 3 (Sjögren’s disease) ⬇️
Aurinia - Lupkynis (calcineurin inhibitor) / Phase 3 (LN) ⬇️
Novartis - Rapcabtagene autoleucel (CD19 CAR-T) / Phase 2 (IIL, SSc) ⬇️
UCB - Bimzelx (anti-IL-17A/F mAb) versus Skyrizi (anti-IL23 mAb) / Phase 3b (PsA)
Potentially best-in-disease
This was arguably the most talked-about oral presentation of the congress. It provided a direct, head-to-head clinical evaluation pitting the dual IL-17A/F mechanism against the dominant IL-23 path in active psoriatic arthritis (PsA).
Indication: Psoriatic Arthritis (PsA) is a prominent, highly heterogeneous type of spondyloarthritis that occurs in about 30% of patients with psoriasis. It uniquely blends this spinal and entheseal inflammation with painful peripheral joint swelling (often causing “sausage digits” or dactylitis), skin lesions, and nail dystrophy, driven by an overactive immune response typically mediated by the IL-17/IL-23 cytokine pathways.
Mechanism: The trial represents a strategic mechanistic clash between upstream cytokine inhibition (IL-23) and direct, downstream dual-isoform blockade (IL-17A/F).
Bimzelx (bimekizumab): A humanized IgG1 monoclonal antibody that dual-inhibits both interleukin-17A (IL-17A) and interleukin-17F (IL-17F). IL-17A and IL-17F are homodimers (and IL-17A/F heterodimers) that share structural homology and cooperate synergistically to drive the tissue inflammation, enthesitis, and new bone formation characteristic of spondyloarthritis. Dual blockade achieves a deeper suppression of downstream inflammatory genes than blocking IL-17A alone.
Skyrizi (risankizumab): A humanized IgG1 monoclonal antibody that selectively targets the p19 subunit of interleukin-23 (IL-23). By inhibiting IL-23, it prevents the differentiation, proliferation, and survival of pathogenic Th17 cells upstream, thereby reducing the production of IL-17 and other downstream effectors.
Trial design: The Phase 3b BE BOLD Trial was a randomized, multicenter, open-label, assessor-blinded (PROBE design), active-controlled, head-to-head superiority trial in adults with active, moderate-to-severe Psoriatic Arthritis (PsA) who met the CASPAR criteria, had ≥3 tender and ≥3 swollen joints, and had an inadequate response or intolerance to at least one conventional synthetic DMARD (csDMARD) or anti-TNF therapy. Patients were randomized to either Bimzelx or Skyrizi. The primary endpoint was the proportion of patients achieving an ACR50 response at Week 16 (signifying a 50% improvement in core American College of Rheumatology joint disease metrics).
Data (press release):
Efficacy: The oral presentation (Abstract LB0001) demonstrated distinct superiority for the dual IL-17A/F approach in rapid and deep joint responses. Bimzelx achieved a statistically significant and clinically meaningful superiority over Skyrizi, with 49.1% of patients achieving ACR50 at Week 16 compared to 38.4% in the Skyrizi arm (p < 0.05). A dramatic kinetic separation was observed as early as Month 1. At Week 4, 19.9% of Bimzelx patients had already reached an ACR50 response, versus just 7.2% of Skyrizi patients. While both agents are highly effective skin clearance tools, Bimzelx held the edge at Week 16 with 53.4% of patients achieving completely clear skin (PASI 100) compared to 46.6% for Skyrizi.
Safety: No new safety signals were observed. Treatment-emergent adverse events (TEAEs) were broadly comparable. As expected with the class, mild-to-moderate oral candidiasis (thrush) rates were numerically higher in the Bimzelx arm due to IL-17F’s role in mucosal fungal surveillance, but these rarely led to treatment discontinuation.
Impact:
Potentially Redefining First-Line Biologic Selection: In the highly fragmented PsA market, these data challenge the growing adoption of IL-23 inhibitors as the default first-line biologic over older anti-TNFs. Demonstrating superior joint efficacy against the market-leading IL-23 (Skyrizi) establishes Bimzelx as an ultra-potent option when rapid joint stabilization is a priority.
The “4-for-4” Moniker: With this readout, UCB has achieved a clean sweep of head-to-head trials, having previously demonstrated Bimzelx‘s superiority over Humira (adalimumab), Cosentyx (secukinumab), and Stelara (ustekinumab). This robust comparative data package strengthens its position in commercial formulary negotiations and clinical guidelines.
Next steps: The immediate next step for the BE BOLD trial is the readout of longer-term data (Weeks 24 and 52) to evaluate whether Bimzelx‘s early joint and skin superiority maintains its separation or if Skyrizi catches up once steady-state dosing is sustained out to a year. Publication of detailed secondary metrics focusing specifically on enthesitis (inflammation where bone meets tendon) and dactylitis (sausage digits) resolution, two highly debilitating manifestations of SpA where IL-17 biology plays a critical localized role.
Biogen - Dapirolizumab (anti-CD40L mAb) / Phase 3 (SLE)
Potentially first-in-class
Systemic Lupus Erythematosus (SLE) has historically been a graveyard for clinical assets. Simultaneous with its presentation at EULAR 2026, full data from the PHOENYCS GO trial evaluating this novel, Fc-free anti-CD40L pegylated Fab fragment was published in The Lancet.
Indication: Systemic Lupus Erythematosus (SLE) is a chronic, heterogeneous autoimmune disease driven by a profound loss of immune tolerance to self-antigens. Its pathophysiology centers on defective clearance of apoptotic cellular debris, which leads to an accumulation of nuclear antigens; these self-antigens trigger B-cell hyperactivation and the subsequent production of pathogenic antinuclear autoantibodies (ANAs) and anti-double-stranded DNA (anti-dsDNA). These autoantibodies bind to self-antigens and form immune complexes that deposit in vital tissues—such as the kidneys, skin, joints, and blood vessels—activating the complement cascade and driving relentless, type I interferon (IFN)-mediated tissue inflammation and organ damage. The clinical standard of care (SOC) aims to suppress this inflammation, prevent acute flares, and minimize long-term organ toxicity. The foundational therapeutic backbone for all patients is hydroxychloroquine (an antimalarials drug), which is layered with systemic glucocorticoids (corticosteroids) for acute flare management, alongside traditional immunosuppressants like mycophenolate mofetil (MMF), methotrexate, or azathioprine for moderate-to-severe or organ-threatening disease. For patients with inadequate disease control, targeted biologic therapies—specifically belimumab (an anti-BAFF monoclonal antibody) or anifrolumab (an anti-type I interferon receptor antibody)—are integrated into the regimen to facilitate steroid tapering and protect long-term organ function.
Mechanism: Dapirolizumab pegol is a novel, investigational humanized Fc-free polyethylene glycol (PEG)-conjugated antigen-binding (Fab’) fragment that selectively targets and inhibits CD40 Ligand (CD40L / CD154). CD40L is primarily expressed on activated T cells and platelets, binding to CD40 on B cells and antigen-presenting cells (APCs). Inhibiting this costimulatory pathway effectively disrupts pathogenic T-cell/B-cell crosstalk, which downregulates B-cell activation, diminishes type I interferon (IFN) secretion, and limits downstream autoantibody production. Historical attempts by the industry to target CD40L were completely halted due to severe thromboembolic (blood clotting) safety issues. Those older antibodies possessed an intact Fc region that bound to platelets, triggering aggregation. Because DZP is Fc-free, it is designed to avoid binding platelet Fc-gamma-RIIa receptors, aiming to mitigate the mechanism of vascular toxicity seen in historical candidates.
Trial design: The Phase 3 PHOENYCS GO Trial was designed to evaluate the safety and efficacy of dapirolizumab added to standard of care (SOC) in Systemic Lupus Erythematosus (SLE). The trial recruited 321 adults with moderately-to-severely active SLE despite receiving standard-of-care (SOC) background therapy (antimalarials, corticosteroids, and/or immunosuppressants). Patients were randomized in a 2:1 ratio to receive either dapirolizumab pegol plus SOC (n=213) or placebo plus SOC (n=108). The primary endpoint was the proportion of patients achieving a BICLA response at Week 48 (British Isles Lupus Assessment Group-based Composite Lupus Assessment).
Data (Biogen press release, UCB press release, Lancet paper):
Efficacy: The drug demonstrated broad, statistically significant improvements across both clinical endpoints and serological markers. At Week 48, 50% (103/208) of patients treated with dapirolizumab pegol achieved a BICLA response compared to 35% (37/106) in the placebo group (p = 0.011). Sapirolizumab significantly reduced the frequency of severe flares and enabled the tapering of steroids. Among patients entering the trial on a baseline corticosteroid dose >7.5 mg/day (prednisone equivalent), a significantly greater proportion in the active arm successfully tapered to ≤7.5 mg/day while maintaining absolute disease control through Week 48. For patients with abnormal baselines, DZP led to significant reductions in anti-dsDNA autoantibodies alongside meaningful increases in complement proteins C3 and C4, validating the drug’s upstream mechanism.
Safety: Treatment-emergent adverse events (TEAEs) were slightly higher in the active arm (82.6% vs 75.0%), but crucially, serious TEAEs were less frequent with DZP than with placebo (10% vs 14.8%). Discontinuations due to adverse events remained low across both groups (4.7% vs 3.7%), and no thromboembolic signal was observed.
Impact:
Graveyard Defied: Systemic lupus is notoriously heterogeneous, making successful Phase 3 assets incredibly rare. This successful readout validates CD40L inhibition as a viable, safe, and highly efficacious pathway when engineered correctly.
Paradigm Shift Away from Steroids: Showing that patients can successfully taper down to safe steroid thresholds (≤ 7.5 mg/day) without triggering flares addresses one of the single largest unmet needs in lupus management, preventing cumulative, drug-induced organ damage.
Next steps: Per standard FDA requirements for broad SLE indications, Biogen and UCB are currently executing a second, parallel confirmatory Phase 3 registrational trial titled PHOENYCS FLY. The positive PHOENYCS GO data will serve as the anchor for global regulatory dossiers (BLA/MAA), which will be filed sequentially as soon as the confirmatory FLY data reads out and solidifies the safety-to-efficacy matrix.
Zenas BioPharma - Obexelimab (CD19 x FcγRIIb mAb) / Phase 3 (IgG4-RD)
Potentially first-in-disease Novel mechanism
The presentation of the Phase 3 INDIGO trial at the EULAR 2026 Congress in London. It was simultaneously published in The New England Journal of Medicine (NEJM). This data marks a significant milestone for Zenas BioPharma and the management of IgG4-Related Disease (IgG4-RD).
Indication: IgG4-Related Disease (IgG4-RD) is a chronic, immune-mediated fibro-inflammatory condition characterized by a unique immune cascade where activated T-follicular helper (TFH) cells and type 2 helper T (TH2) cells drive the continuous hyper-activation and expansion of B-cell lineages, specifically IgG4-positive plasmablasts and plasma cells. These cells infiltrate vital organs (such as the pancreas, salivary glands, retroperitoneum, and lymph nodes), triggering severe local tissue swelling and a distinctive “storiform” pattern of progressive fibrosis and obliterative phlebitis that can cause irreversible organ damage or failure. The clinical standard of care (SOC) focuses on quickly arresting this destructive inflammation. Systemic glucocorticoids (corticosteroids) remain the universal first-line treatment to induce rapid clinical remission; however, because IgG4-RD has an exceptionally high relapse rate upon tapering, patients often require long-term, toxic steroid maintenance. To mitigate steroid-induced metabolic and bone toxicities, clinicians integrate traditional steroid-sparing immunosuppressants like mycophenolate mofetil (MMF), azathioprine, or methotrexate.
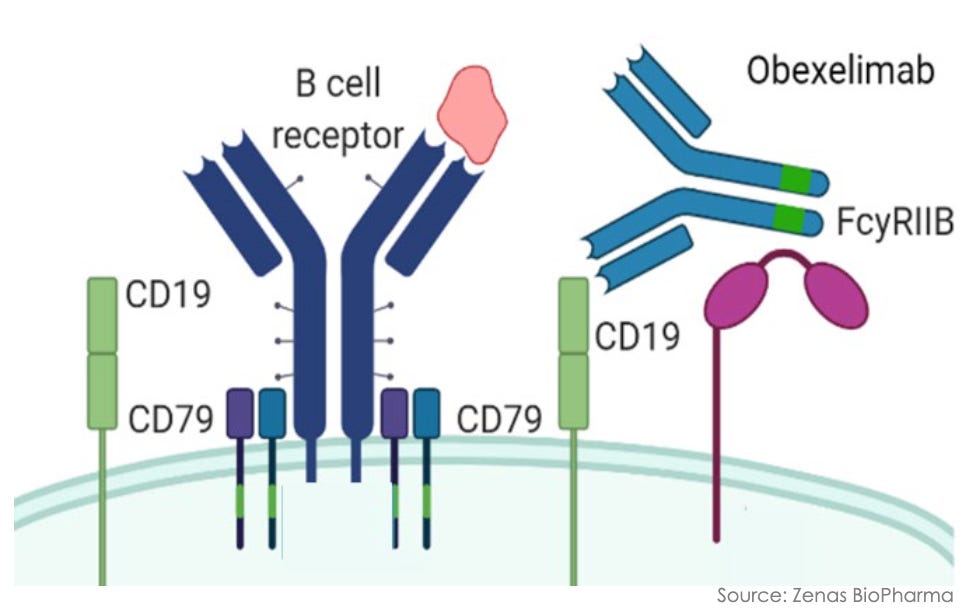
Mechanism: Obexelimab is a first-in-class, humanized, bifunctional monoclonal antibody that selectively targets B-cell lineage cells through a unique inhibitory rather than depleting strategy. The antibody is engineered to simultaneously bind CD19 and the inhibitory Fc receptor Fc-gamma-RIIb (CD32B), both of which are broadly co-expressed across the B-cell lineage (including naive B cells, memory B cells, and plasmablasts). By cross-linking CD19 with Fc-gamma-RIIb, obexelimab co-opts the immune system’s natural negative-feedback loop. This mechanical engagement downregulates B-cell receptor (BCR) signaling, effectively paralyzing and inhibiting B-cell activation, proliferation, and downstream autoantibody secretion. Unlike standard B-cell therapies (e.g., anti-CD20 mAbs like rituximab), obexelimab does not deplete B cells. This leaves the patient’s underlying humoral immune landscape intact, allowing for a rapid return to immune baseline upon drug cessation and mitigating the long-term severe infection risks tied to cellular depletion.
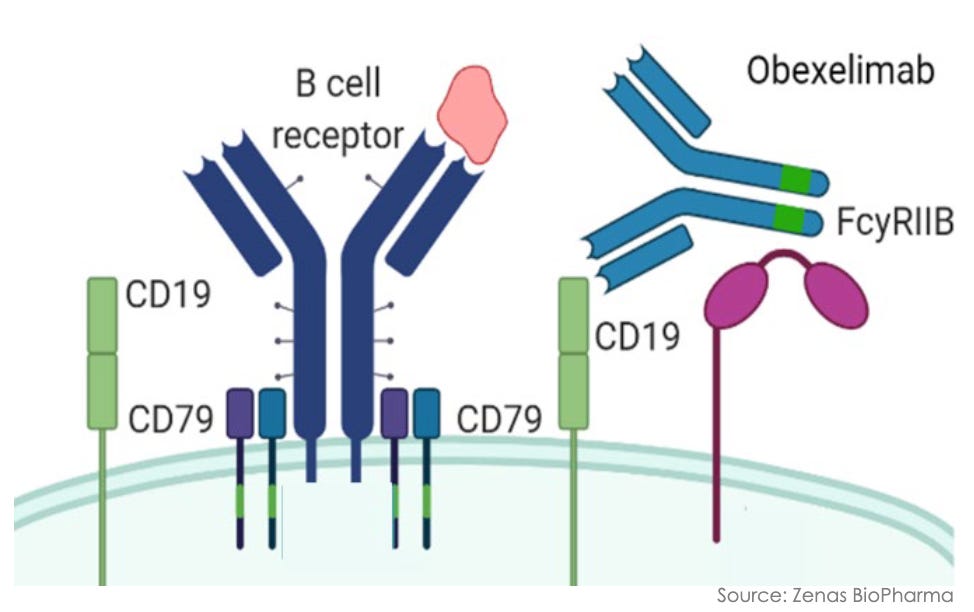
Obexelimab mechanism; Source: Zenas presentation, slide 10 Trial design: The Phase 3 INDIGO study stands as the largest randomized, double-blind, placebo-controlled registrational trial ever conducted in IgG4-RD. The trial recruited 194 adults with newly diagnosed or recurrent, active, multi-organ IgG4-RD who met the 2019 ACR/EULAR classification criteria and required immediate systemic glucocorticoid (GC) intervention. Participants were randomized 1:1 to receive either obexelimab (250 mg via subcutaneous injection, weekly) or matching placebo for 52 weeks. To isolate the true therapeutic effect of the biologic, all patients were mandatory-started on an aggressive, protocol-driven background glucocorticoid (prednisone) taper at Day 1, down to 0 mg by Week 8. The primary endpoint was the time to the first IgG4-RD disease flare during the 52-week randomized controlled period, as determined by an independent, blinded Adjudication Committee (AC) and clinical investigators (flares requiring rescue therapy were managed with an open-label glucocorticoid intervention).
Data (press release, NEJM paper):
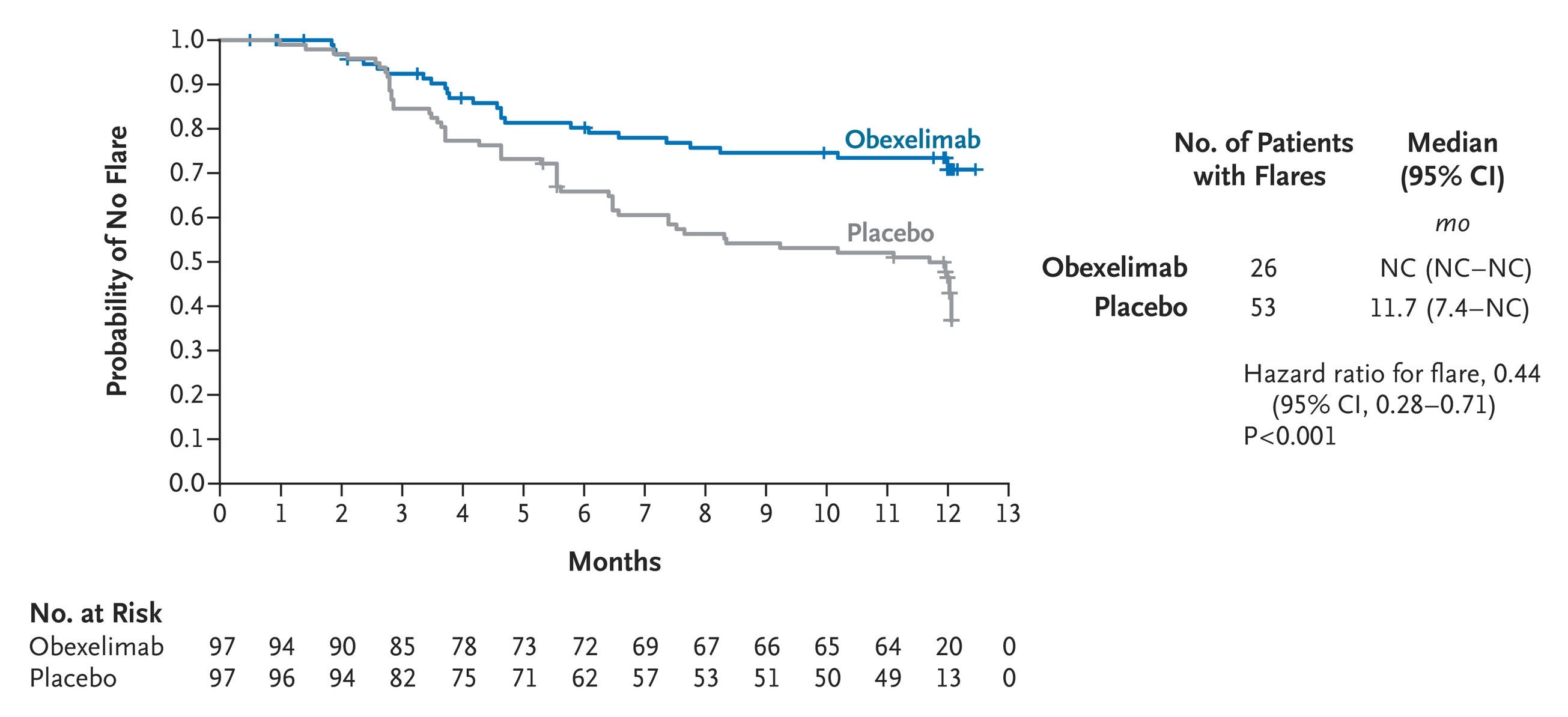
Efficacy: The INDIGO trial achieved a definitive, highly statistically significant clean sweep across its primary and all four key secondary endpoints. Obexelimab demonstrated a striking 56% reduction in the risk of IgG4-RD disease flares over 52 weeks compared to placebo (HR = 0.443, p = 0.0005; see graph below). This significantly prolonged time to flares requiring rescue therapy (p = 0.0001) and dropped the absolute number of rescue-dependent flares (p = 0.0008). A significantly higher proportion of patients in the active arm achieved and maintained complete remission at Week 52 (p = 0.0049). Obexelimab drove a substantial reduction in cumulative glucocorticoid rescue therapy use (p = 0.0042), translating to lower measurable glucocorticoid-induced organ toxicity.
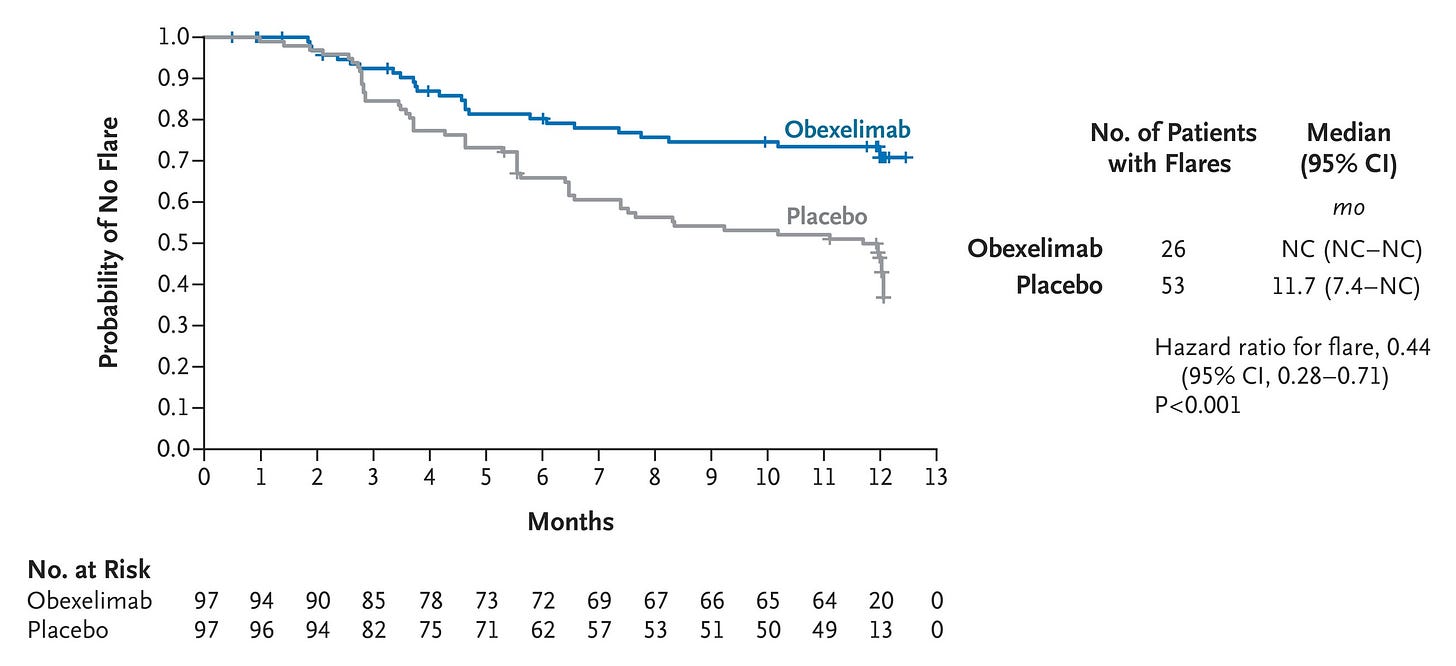
First Adjudicated Flare of IgG4-Related Disease for Which Rescue Therapy Was Required (Intention-to-Treat Population); Source: Della-Torre et al. NEJM (2026), Figure 1 Safety: Overall treatment-emergent adverse events (TEAEs) were balanced (97.9% obexelimab vs. 95.9% placebo). Crucially, Grade ≥3 TEAEs were nearly halved with obexelimab (11.3% vs. 23.7%), and serious adverse events (SAEs) favored the drug at 10.3% vs. 18.6%. Total infections were actually lower in the obexelimab group (53.6% vs. 62.9%), validating the safety profile of an inhibitory versus depleting approach. Injection-site reactions occurred in a modest 3.5% of total active doses, and there were zero deaths in the obexelimab cohort.
Impact:
Establishing the First Standard: There are currently zero FDA-approved targeted therapies for IgG4-RD. Clinicians have been forced to rely on long-term, high-dose steroids (which drive irreversible metabolic and bone damage) or B-cell depletion (rituximab). If approved, some clinical analysts suggest obexelimab could reshape the paradigm as a first-line targeted standard of care.
The Shift to At-Home Care: Rituximab requires resource-intensive, in-clinic intravenous infusions. Obexelimab’s formulation allows for an autonomous, self-administered at-home subcutaneous injection regimen, massively lowering systemic healthcare utilization costs and improving patient quality of life.
Validation of the Non-Depleting B-cell Hypothesis: This robust Phase 3 win serves as a major de-risking event for the broader field of B-cell inhibition, demonstrating that turning B cells “off” can match or exceed the efficacy of killing them, without the associated long-term immunosuppressive safety penalties.
Next steps: Zenas BioPharma executed swiftly around this data, submitting its Biologics License Application (BLA) to the FDA in Q2 2026. The next major catalyst will be the regulatory decision, alongside a planned Marketing Authorization Application (MAA) submission to the European Medicines Agency (EMA) in H2 2026. Patients exiting the 52-week randomized trial have transitioned into a 3-year Open-Label Extension (OLE) study to monitor long-term safety, sustained remission durability, and tissue damage prevention over a multi-year horizon. With the B-cell inhibition mechanism clinically validated in IgG4-RD, Zenas is actively advancing obexelimab into other high-unmet-need autoimmune indications, including an active Phase 2 trial (SunStone) in Primary Sjögren’s Syndrome and other closely aligned rheumatological conditions.
Novartis - Ianalumab (anti-BAFF mAb) / Phase 3 (Sjögren’s disease)
Potentially first-in-disease
The presentation of ianalumab (VAY736) at EULAR 2026 represents a historic milestone for Sjögren’s disease, the second most prevalent rheumatic autoimmune condition which has historically suffered from a complete lack of approved disease-modifying therapies. Building upon the landmark Phase 3 primary endpoints revealed in late 2025, Novartis showcased critical long-term Week 108 interim extension data at EULAR 2026, anchoring its regulatory pathway toward a global launch.
Indication: Sjögren’s disease is a chronic, systemic autoimmune disorder characterized by a progressive loss of immune tolerance that leads to the targeted destruction of exocrine glands, primarily the salivary and lacrimal glands. Its pathophysiology centers on a profound lymphocytic infiltration (chiefly CD4+ T cells and hyperactive B cells) into glandular tissues, driven by the chronic overexpression of B-cell activating factor (BAFF) and type I interferons (IFNs). This chronic inflammatory environment induces secretory epithelial cell dysfunction, leading to severe exocrine hypofunction, tissue fibrosis, and the classic sicca symptoms of xerostomia (profound dry mouth) and keratoconjunctivitis sicca (profound dry eyes), while also predisposing a subset of patients to systemic extraglandular manifestations (e.g., vasculitis, peripheral neuropathy, interstitial lung disease) and a significantly elevated risk of B-cell lymphoma. Because there are historically no approved disease-modifying therapies (DMARDs), the clinical standard of care (SOC) is heavily polarized between localized, reactive palliation and systemic immunosuppression. For sicca features, management relies entirely on symptomatic alleviation via artificial tears, lubricating ophthalmic ointments, topical cyclosporine eye drops, and oral sialagogues like pilocarpine or cevimeline to stimulate residual salivary flow. For patients presenting with severe systemic or extraglandular disease, clinicians escalate to off-label systemic regimens—predominantly utilizing oral glucocorticoids (corticosteroids) for acute flare control, layered with traditional immunosuppressants such as hydroxychloroquine for arthralgias, or methotrexate, azathioprine, and off-label rituximab for severe organ-threatening complications.
Mechanism: Ianalumab is an ADCC-active anti-BAFF antibody that features a unique dual mechanism of action engineered to maximize B-cell eradication and suppression. The antibody is afucosylated (engineered lacking fucose sugar units on its Fc portion). This structural modification dramatically increases its binding affinity to CD16 receptors on Natural Killer (NK) cells. As a result, ianalumab triggers highly potent antibody-dependent cellular cytotoxicity (ADCC), eliminating circulating and, crucially, tissue-infiltrated B cells within the exocrine glands with 40- to 70-fold greater potency than legacy anti-CD20 agents like rituximab. Concurrently, ianalumab binds directly to the B-cell activating factor receptor (BAFF-R). By physically blocking BAFF from attaching to its receptor, it cuts off the essential survival and activation signaling pathways for any remaining or nascent autoreactive B cells, inhibiting their downstream proliferation and autoantibody production.
Trial design: The registrational clinical package consisted of two global, randomized, double-blind, placebo-controlled Phase 3 trials and their subsequent open-label extensions (NEPTUNUS-1 & NEPTUNUS-2 trials). They included a combined enrollment of 779 adult patients across 35 countries with active, moderate-to-severe primary Sjögren’s disease (characterized by systemic disease burden and exocrine glandular dysfunction). Patients were randomized to receive subcutaneous injections of either ianalumab 300 mg monthly, ianalumab every 3 months, or matching placebo for 52 weeks. The primary endpoint was The change from baseline in the ESSDAI score (EULAR Sjögren’s Syndrome Disease Activity Index) at Week 48. The EULAR 2026 presentation focused on long-term efficacy, structural salivary preservation, and safety data capturing patient exposure through an interim Week 108 extension phase.
Data:
Efficacy: Following the successful Week 48 readouts (where monthly ianalumab met the primary endpoint with a statistically significant, rapid, and sustained reduction in ESSDAI score, p = 0.0031), the new Week 108 interim data (Abstract OP0126) added crucial durability metrics. Significant improvements were maintained out to 108 weeks across key patient-reported outcomes, including patient-rated symptom diaries measuring global dryness, chronic widespread pain, and debilitating fatigue (ESSPRI indices). A highly impactful sub-analysis showed that in patients who maintained a baseline stimulated salivary flow rate of ≥0.4 mL/min, ianalumab therapy successfully halted further deterioration of glandular function and reduced objective oral dryness over the 108-week horizon compared to historical placebos.
Safety: Extended exposure did not yield any new safety signals. Total incidence of treatment-emergent adverse events (TEAEs) and serious infections remained thoroughly comparable to the placebo baseline, validating the targeted safety window of BAFF-R blockade over non-selective immunosuppression.
Impact:
Potentially First-in-disease: Having zero approved competitors grants Novartis an open runway. With an estimated 50% of the Sjögren’s population currently going undiagnosed due to limited historical treatment options. Like lupus, Sjögren’s disease has been notoriously difficult to treat, causing decades of failed clinical trials. Ianalumab stands as the first targeted biologic in history to hit its primary endpoints in Phase 3 trials for this indication.
Disease-Modifying Potential: Rather than just masking symptoms with eye drops or artificial saliva, ianalumab treats the underlying pathology. This shifts the clinical standard from reactive, symptomatic palliation to active, long-term organ and tissue preservation.
Next steps: Backed by an FDA Breakthrough Therapy Designation awarded in January 2026, Novartis has initiated global regulatory filings, with a target FDA approval decision and subsequent European Medicines Agency (EMA) submission timelines running throughout late 2026 and early 2027. Continued monitoring of the NEPTUNUS open-label extension cohorts out to their final protocol endpoints to evaluate long-term safety, immunoglobulin recovery kinetics, and potential impacts on reducing lymphoma risk (Sjögren’s patients carry a 5% to 10% lifetime risk of developing B-cell lymphoma). Utilizing the validated dual BAFF-R/ADCC backbone, Novartis is actively progressing ianalumab across a wide, corporate-wide multi-indication footprint, including ongoing trials in Systemic Lupus Erythematosus (Phase 2 data also highlighted at EULAR 2026), Lupus Nephritis, Immune Thrombocytopenia (ITP), and Diffuse Cutaneous Systemic Sclerosis.
Aurinia - Lupkynis (calcineurin inhibitor) / Phase 3 (LN)
At EULAR 2026, Aurinia Pharmaceuticals presented a highly impactful, new long-term time-to-event safety and efficacy analysis from its landmark AURORA 1 Phase 3 study of Lupkynis (voclosporin). While Lupkynis is already FDA-approved, this new data package significantly elevates its clinical profile by demonstrating a massive reduction in “hard” clinical endpoints, such as kidney failure and death, solidifying its competitive positioning in the crowded lupus nephritis (LN) landscape.
Indication: Lupus Nephritis (LN) is a severe, organ-threatening manifestation of SLE occurring in up to 50% of patients, driven by an autoimmune assault where circulating anti-dsDNA autoantibodies and nuclear antigens form immune complexes that deposit directly within the renal glomeruli. This intrarenal deposition activates the complement cascade, triggering a massive influx of inflammatory cells, podocyte injury, and severe structural damage across the glomerular basement membrane. If left unchecked, this localized inflammatory cascade progresses to glomerulosclerosis, interstitial fibrosis, and irreversible chronic kidney disease (CKD) or end-stage renal disease (ESRD). The clinical standard of care (SOC) aims to rapidly halt this renal inflammation to maximize nephron preservation while minimizing drug toxicity. Standard therapy is divided into an intensive induction phase to achieve complete renal response—historically utilizing a heavy backbone of systemic high-dose glucocorticoids (corticosteroids) paired with mycophenolate mofetil (MMF) or intravenous cyclophosphamide—followed by a lower-dose maintenance phase using MMF or azathioprine to prevent relapses. Modern clinical guidelines now heavily favor upfront multi-target or triple-therapy regimens for active LN, layering advanced targeted biologics like belimumab (anti-BAFF mAb) or adding a next-generation oral calcineurin inhibitor like voclosporin (Lupkynis) to the background MMF/steroid regimen to accelerate proteinuria reduction and facilitate aggressive, organ-protecting steroid tapering.
Mechanism: Lupkynis (voclosporin) is a next-generation, structurally modified oral calcineurin inhibitor (CNI). Compared to older CNIs like cyclosporine or tacrolimus, Lupkynis exhibits a predictable pharmacokinetic-pharmacodynamic relationship. This eliminates the operational burden of routine therapeutic drug monitoring (TDM) and minimizes standard CNI toxicities (such as severe lipid or glucose metabolic derangements).
Trial design: The data presented at EULAR 2026 evaluated a post-hoc, time-to-event clinical analysis of the pivotal AURORA 1 Phase 3 trial. The trial recruited 356 randomized adult patients with active, biopsy-proven Class III, IV, or V Lupus Nephritis (178 on Lupkynis, 178 on placebo). All patients received a multi-target background standard of care consisting of mycophenolate mofetil (MMF) and an aggressive, low-dose oral corticosteroid taper. Patients were randomized 1:1 to receive either Lupkynis (23.7 mg twice daily) or matching placebo layered onto the background SOC for 52 weeks. A rigorous composite time-to-event endpoint measuring the safety population’s risk of a renal-related event or death, tracking components including treatment failure, worsening proteinuria, and all-cause mortality.
Data (press release, poster):
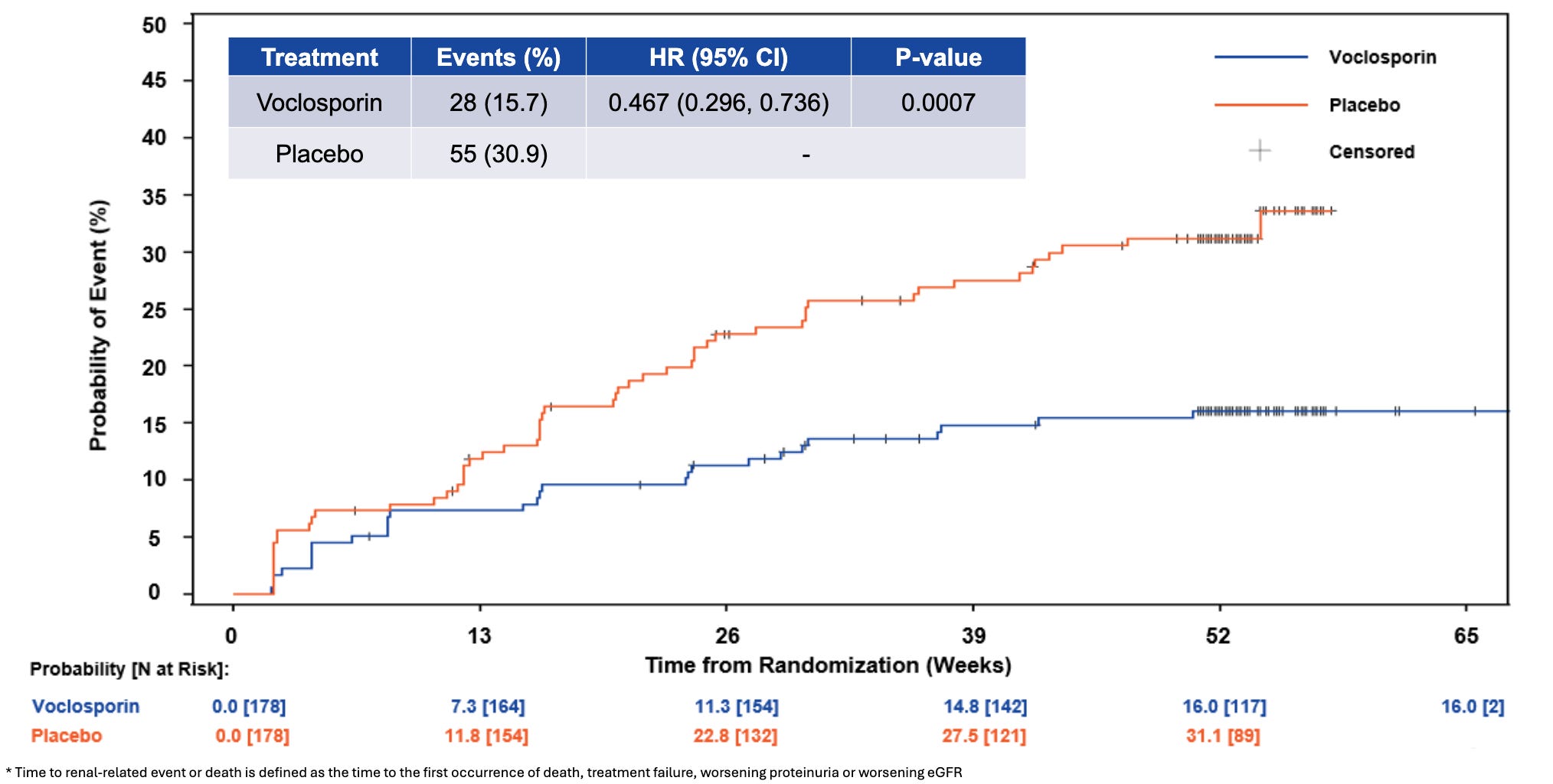
Efficacy: The newly presented analysis (led by Dr. Amit Saxena) demonstrated that adding Lupkynis to MMF and steroids dramatically alters long-term renal outcomes. Treatment with Lupkynis was associated with a highly statistically significant 53% reduction in the risk of a renal-related event or death compared to placebo (HR = 0.47, p = 0.0007). Lupkynis drove a 78% reduction in the risk of progressive proteinuria (HR = 0.22, p < 0.0001), highlighting immediate, sustained podocyte protection. Associated with a 55% reduction in the risk of clinical treatment failure (HR = 0.45, p = 0.0062). Demonstrated a strong trend favoring the active arm with an 81% reduction in the risk of death (HR = 0.19, p = 0.0929). The presentation reiterated that Lupkynis remains the only oral option to show statistically significant improvements in Complete Renal Response (CRR) in as short as 6 months within a randomized Phase 3 setting.
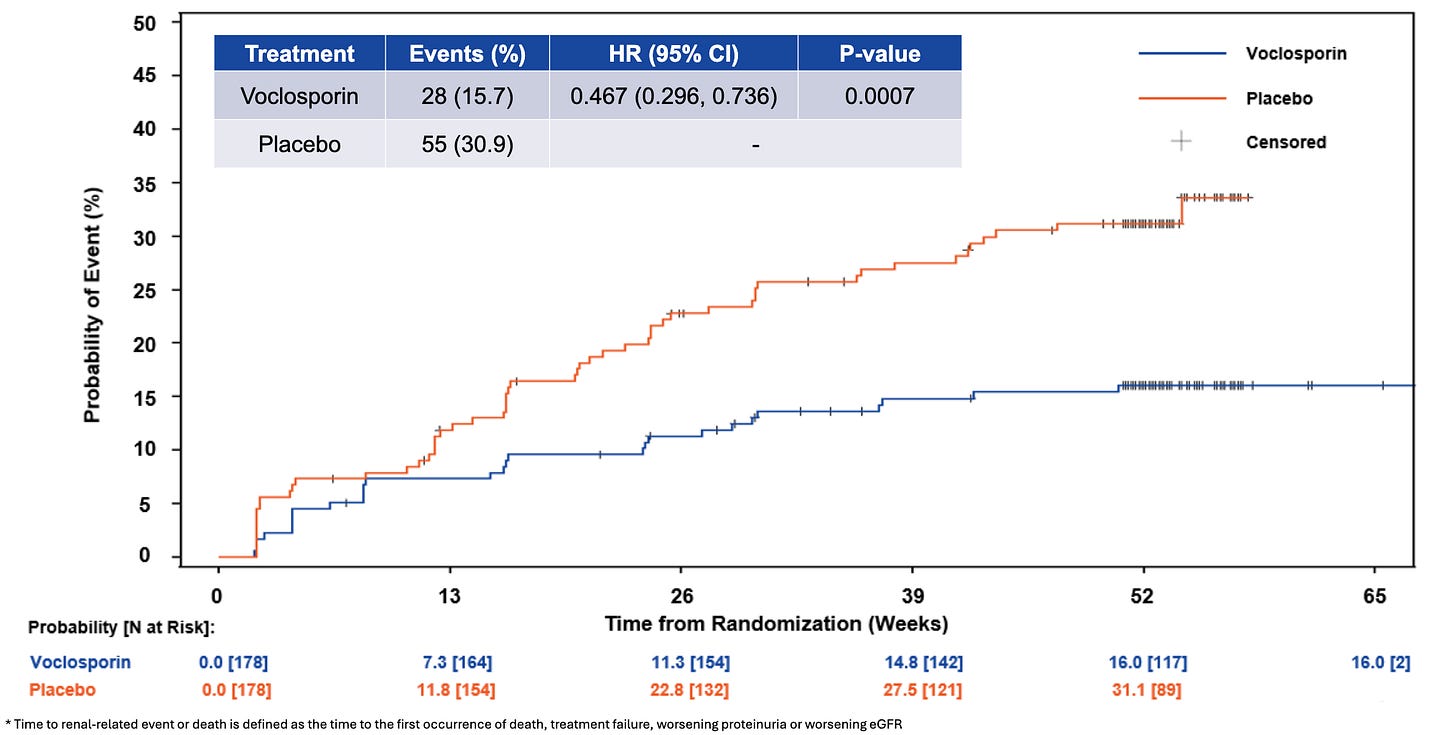
Time to Renal-Related Event or Death; Source: Aurinia poster, Figure 4 Safety: The FDA-approved label for Lupkynis (voclosporin) carries a Boxed Warning regarding an increased risk for developing serious infections (including bacterial, viral, fungal, and opportunistic infections that can lead to hospitalization or death) and malignancies (such as lymphoma and skin cancers) due to its potent immunosuppressive nature. According to its clinical safety profile, the most common adverse reactions (≥3%) include decreased estimated glomerular filtration rate (eGFR), hypertension, diarrhea, headache, anemia, cough, urinary tract infections, and abdominal pain. Additionally, because it is a calcineurin inhibitor, Lupkynis can cause acute, dose-dependent nephrotoxicity and neurotoxicity (such as tremors, seizures, or altered mental status), can prolong the QT interval when combined with other QT-prolonging drugs, and may cause hyperkalemia. It is also contraindicated for co-administration with strong CYP3A4 inhibitors (like ketoconazole) due to a substantial increase in drug exposure.
Impact:
Payer and Provider Leverage: Showing a 53% reduction in hard endpoints like renal degradation and death shifts the clinical conversation from a surrogate biomarker (proteinuria reduction) to concrete organ preservation, which is vital for winning commercial payer coverage and formulary placement.
Defending Market Share: The LN space is intensely competitive, with Lupkynis going toe-to-toe with GSK’s intravenously/subcutaneously infused Benlysta (belimumab) and Roche/Genentech’s newly reading-out Phase 3 anti-CD20 mAb obinutuzumab (REGENCY trial). Confirming that an entirely oral multi-target regimen prevents long-term renal failure strengthens Lupkynis‘s foothold among community nephrologists and rheumatologists.
Next steps: Aurinia is pushing for these long-term hard outcome data points to be formally incorporated into upcoming EULAR, ACR, and KDIGO clinical management guidelines for lupus nephritis, positioning early oral CNI introduction as a gold standard. Growing the Lupkynis franchise allows Aurinia to fund and advance its second preclinical/clinical pipeline asset, aritinercept (a dual BAFF/APRIL inhibitor aimed at broader autoimmune diseases) reducing the company’s reliance on a single marketed product.
Novartis - Rapcabtagene autoleucel (CD19 CAR-T) / Phase 2 (IIL, SSc)
At EULAR 2026, Novartis presented highly anticipated data from its cell therapy pipeline, highlighting its autologous CD19-directed CAR-T cell therapy, rapcabtagene autoleucel (rap-cel, formerly YTB323). The spotlight fell on an oral presentation detailing preliminary analysis from the AUTOGRAPH-IIM and AUTOGRAPH-SSc trials, marking a push to transition oncology-born CAR-T architectures into severe, life-threatening autoimmune diseases.
Indication: Idiopathic Inflammatory Myopathies (IIM / Myositis) and Systemic Sclerosis (SSc / Scleroderma) are severe, chronic systemic autoimmune diseases characterized by profound immune dysregulation that leads to progressive organ dysfunction and tissue scarring. In IIM, the pathophysiology centers on cellular and humoral autoantibody attacks (such as anti-synthetase or myositis-specific antibodies) directly targeting skeletal muscle fibers and small vasculature, inducing muscle cell necrosis, severe symmetric proximal muscle weakness, and extra-muscular inflammation of the skin, joints, and lungs. In SSc, the pathophysiology involves a devastating triad of microvascular injury, intense autoimmune hyperactivation (marked by anti-Scl-70 or anti-centromere antibodies), and massive, uncontrolled overproduction of extracellular matrix proteins by myofibroblasts, causing a characteristic thickening and hardening of the skin (fibrosis) alongside progressive, life-threatening scarring of internal organs like the lungs (interstitial lung disease), heart, and gastrointestinal tract. The clinical standard of care (SOC) for both conditions heavily prioritizes aggressively suppressing active immunologic inflammation to arrest downstream tissue degradation. For IIM, treatment begins with high-dose systemic glucocorticoids (corticosteroids), rapidly layered with traditional steroid-sparing immunosuppressive DMARDs such as methotrexate, azathioprine, or mycophenolate mofetil (MMF), and escalated to intravenous immunoglobulin (IVIG) or rituximab for severe or refractory cases. For SSc, management is highly organ-specific due to the lack of a universal disease-modifying cure; it utilizes MMF or cyclophosphamide to combat progressive interstitial lung disease, often combined with the antifibrotic tyrosine kinase inhibitor nintedanib, while peripheral vascular complications like Raynaud’s phenomenon are managed with vasodilators (calcium channel blockers, PDE-5 inhibitors) and hypertensive renal crises are aggressively treated with ACE inhibitors.
Mechanism: Rapcabtagene autoleucel is an autologous, CD19-targeted Chimeric Antigen Receptor (CAR) T-cell therapy, engineered utilizing Novartis’s proprietary T-Charge rapid manufacturing platform. They seek out and deeply deplete pathogenic B-cell populations not just in the peripheral blood, but deep within affected target tissues. As the CAR-T cells contract, a completely “naive” and un-primed B-cell population repopulates from the bone marrow, effectively resetting the patient’s immune tolerance. Traditional oncology CAR-T manufacturing takes weeks, leading to cell exhaustion. Novartis’s T-Charge platform reduces the ex vivo culture time to less than two days.
Trial design: The Phase 2 data presented at EULAR emerged from two parallel, open-label, multi-center, single-arm clinical studies evaluating the feasibility, safety, and early efficacy of rap-cel. AUTOGRAPH-IIM enrolled adult patients with Severe Refractory Idiopathic Inflammatory Myopathies (IIM) (including polymyositis, dermatomyositis, and anti-synthetase syndrome) who displayed active muscle weakness and inadequate responses to standard high-dose corticosteroids and multiple traditional immunosuppressive or biologic DMARDs. AUTOGRAPH-SSc enrolled adult patients with active, severe Diffuse Cutaneous Systemic Sclerosis (dcSSc / scleroderma) characterized by rapidly progressive skin fibrosis and a high risk of or existing internal organ involvement (such as interstitial lung disease). Patients underwent a standard 3-day lymphodepletion conditioning regimen consisting of fludarabine and cyclophosphamide, followed immediately by a single intravenous infusion of rap-cel.
Data:
Efficacy: The oral abstract presentation (Abstract OP0077) provided a critical preliminary peek into the expansion kinetics, tissue clearance, and safety profiles of rap-cel in these non-oncology populations. Rap-cel demonstrated a robust, predictable expansion wave, tracking a distinct in vivo peak within 14 to 21 days post-infusion, followed by gradual contraction. This kinetic wave triggered immediate, complete peripheral B-cell aplasia alongside rapid clearance of baseline autoimmune serum biomarkers. In IIM, patients demonstrated early, meaningful improvements in Total Improvement Scores (TIS), characterized by stabilization or recovery of proximal muscle strength and reductions in muscle enzyme levels (e.g., creatine kinase). In SSc, early multi-domain stabilization was observed, highlighted by reductions in the Modified Rodnan Skin Score (mRSS) measuring skin tightness, and early stabilization of pulmonary metrics (Forced Vital Capacity) in individuals presenting with baseline interstitial lung disease.
Safety: Crucially for an autoimmune population, rap-cel exhibited a manageable safety profile. Cytokine Release Syndrome (CRS) was overwhelmingly restricted to mild-to-moderate (Grade 1 or 2) transient events, resolving rapidly with standard protocols (such as tocilizumab). Immune Effector Cell-Associated Neurotoxicity Syndrome (ICANS) and unexpected off-target toxicities were remarkably rare or absent, confirming that the ultra-low cell dose facilitated by the T-Charge™ platform acts as an effective safety buffer.
Impact:
Moving Past Temporary Palliation: Current standard therapies for IIM and SSc merely dampen inflammation or slow tissue scarring. Early cohort data highlights the investigational potential of a functional cure or long-term drug-free remission via an intensive, one-time treatment intervention.
Strategic Differentiation: The data positions Novartis at the forefront of the highly competitive “Autoimmune CAR-T Race” (see our CAR-T, Part 3 piece for more on the landscape), keeping stride with rival academic data (such as the landmark Erlangen cohorts) and specialized biotechs (like Kyverna’s KYV-101 or Cabaletta’s CABA-201).
The Long-Term Durability Question: Independent registry meta-analyses presented during the same sessions (Abstract OP0076) noted that across all historical global autoimmune CAR-T recipients, a minority (~12%) eventually experienced clinical relapses (median 13 months), which occurred more frequently in IIM than in SLE or SSc. Novartis’s data acts as a baseline to prove whether the unique, stem-like properties of T-Charge manufactured cells provide superior long-term remission durability over standard manufacturing methods.
Next steps: Novartis is scaling recruitment for its global, randomized, open-label, active-controlled Phase 2 trial (NCT06665256), which aims to randomize approximately 123 patients to directly test rapcabtagene autoleucel against investigator’s choice of standard background comparator therapies in severe IIM. Concurrently, Novartis presented 24-month long-term tracking data in severe refractory SLE patients treated with rap-cel (Abstract POS0303), establishing a blueprint for the multi-year long-term follow-up (LTFU) protocols mandatory for the IIM and SSc arms to observe the precise timelines of B-cell reconstitution. Refining the precise, minimal dose requirements and lymphodepletion intensities needed to guarantee absolute tissue-level B-cell clearance while minimizing the neutropenic infection window for fragile, multi-organ compromised scleroderma patients.
To contact us, please send us an email at biotechreadout@gmail.com
Disclaimers
Investigational Status Disclaimer
The therapeutic candidates discussed in this newsletter are currently in clinical development and have not been approved for commercial sale by the U.S. Food and Drug Administration (FDA), the European Medicines Agency (EMA), or other global regulatory authorities. Their safety and efficacy have not been established. References to pipeline products and ongoing clinical trials involve significant risks and uncertainties. Statements regarding the potential safety, potency, or efficacy of investigational drugs reflect current hypotheses and are not a guarantee of future performance or regulatory clearance. The outcome of clinical trials is inherently unpredictable, and clinical results from earlier stages may not be predictive of results in later, larger-scale trials.
No Medical Advice Disclaimer
This newsletter is for informational and educational purposes only. The content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or another qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read in this publication.
No Patient-Provider Relationship Disclaimer
The information provided in this newsletter is for educational and analytical purposes only. Receipt of this information, or any interaction with this content, does not create a physician-patient, pharmacist-patient, or any other professional-provider relationship between you and the authors or publishers. This newsletter should not be used as a substitute for a personal consultation with a qualified healthcare professional.
Forward-Looking Statements Disclaimer
This newsletter contains “forward-looking statements” regarding future events, including clinical trial timing, regulatory milestones, and projected market performance. These statements are based on current expectations and assumptions that are subject to significant risks and uncertainties. Actual results may differ materially from those expressed or implied. We undertake no obligation to update these statements as a result of new information or future developments.
Third-Party Links & Content Disclaimer
This newsletter contains links to third-party websites, including clinical trial registries and corporate presentations. Biotech Readout does not endorse, guarantee, or assume responsibility for the accuracy or reliability of any information offered by third-party providers.
Errors and Omissions Disclaimer
While we strive for technical accuracy, the information in this newsletter is provided on an “as is” basis with no guarantees of completeness, accuracy, or timeliness. Biotech Readout assumes no liability for any errors or omissions in the content of this publication.
Non-Endorsement Disclaimer
Any reference to specific commercial products, processes, or services by trade name, trademark, or manufacturer does not constitute or imply an endorsement or recommendation by the author. All trademarks are the property of their respective owners.
No Investment Advice Disclaimer
This newsletter is for informational purposes only and does not constitute financial, investment, or legal advice. The author is not a registered investment advisor. You should consult with a professional financial advisor before making any investment decisions. The biotechnology sector is highly volatile; past performance is not indicative of future results.
Conflict of Interest Disclaimer
The author of this newsletter maintains a position of independence. At the time of publication, the author holds no direct financial interest, equity, or options in any of the companies mentioned in this report. No compensation has been received from any third party to feature or analyze specific therapeutic candidates or corporate entities.