
ASCO 2026 Awards
Major survival wins in pancreatic, breast, and skin cancer

Disclaimer: This newsletter is for educational and informational purposes only and does not constitute medical, investment, or financial advice, nor does it establish a provider-patient relationship. Content may include forward-looking statements and discussions of investigational therapeutic candidates that are not FDA/EMA approved; their safety and efficacy remain unestablished and clinical outcomes are unpredictable. While we strive for accuracy, all information is provided as is without guarantees. This newsletter is independent, and the author holds no financial positions in the companies mentioned nor receives third-party compensation for this coverage. Please find a complete version of our disclaimers at the bottom of this article and on our About page.

Press ⬇️ to go down to a section and ⬆️ to go back up to the Table of Contents
Table of Contents
The 2026 American Society of Clinical Oncology (ASCO) Annual Meeting, which wrapped up this past week (May 29 - June 2) in Chicago, centered on the theme “The Science and Practice of Translation: Improving Cancer Outcomes Worldwide.” With over 44,000 global oncology professionals in attendance, the meeting delivered several massive, potentially practice-changing clinical datasets.
This week, I present the ASCO 2026 Awards, which are based on my personal opinion of patient impact and strictly for fun:
GOLD Revolution Medicines - Daraxonrasib (pan-RAS inhibitor) / Phase 3 (mPDAC) ⬇️
SILVER Celcuity - Gedatolisib (Pan-PI3K/mTOR inhibitor) / Phase 3 (HR+ HER2- mBC) ⬇️
BRONZE Ideaya - Darovasertib (PKC inhibitor) / Phase 2/3 (MUM) ⬇️
My Honorable Mention includes the emerging field of in vivo CAR-T therapy, which stole the spotlight at ASCO 2026 and is a rapidly growing theme that we covered in CAR-T, Part 2. By programming T-cells directly inside the patient’s body, these therapies circumvent apheresis, weeks-long ex vivo manufacturing delays, and the toxicities of lymphodepleting chemotherapy.
Honorable Mention Kelonia / Eli Lilly - KLN-1010 (in vivo BCMA CAR-T) / Phase 1/2 (MM) ⬇️

GOLD Revolution Medicines - Daraxonrasib (pan-RAS inhibitor) / Phase 3 (mPDAC)
The Phase 3 RASolute 302 trial data for daraxonrasib (RMC-6236) presented at ASCO 2026 and concurrently published in the New England Journal of Medicine represents a historic moment for a disease context that has been notoriously refractive to targeted therapies.
Indication: Metastatic pancreatic ductal adenocarcinoma (mPDAC) is a highly aggressive malignancy driven by a dense, immunosuppressive desmoplastic stroma and near-ubiquitous driver mutations, most notably in the KRAS oncogene (>90% of cases), alongside inactivating mutations in tumor suppressors like TP53, CDKN2A, and SMAD4. This rich fibroinflammatory microenvironment creates high interstitial fluid pressure and profound hypoxia, severely impairing drug delivery and rendering the tumor largely resistant to standard immunotherapy. Consequently, systemic therapy relies on combination cytotoxic chemotherapy backbones. The frontline standard of care is determined by patient performance status, typically utilizing either FOLFIRINOX (or modified FOLFIRINOX) or gemcitabine plus nab-paclitaxel. For the small subset of patients harboring specific genomic alterations, targeted strategies are integrated, such as PARP inhibitors (olaparib) for germline BRCA1/2 mutations or pembrolizumab for MSI-H/dMMR tumors, while second-line regimens generally rotate the alternative chemotherapy backbone (e.g., liposomal irinotecan plus 5-FU/LV following gemcitabine failure) or capitalize on newly emerging targeted options like pan-KRAS inhibitors.
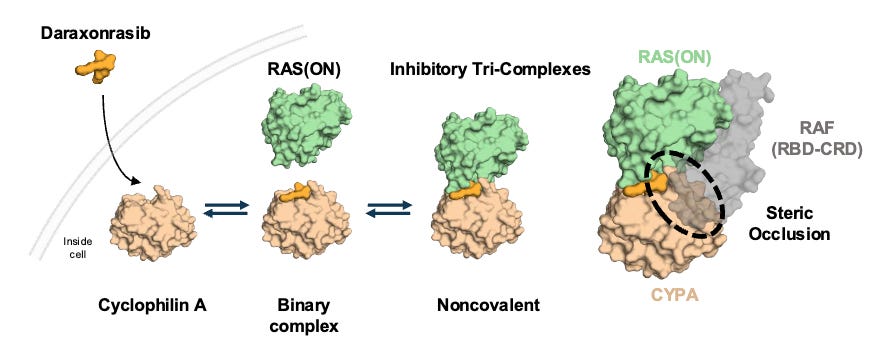
Mechanism: Daraxonrasib is a first-in-class, oral, non-covalent, multi-selective RAS(ON) inhibitor. It acts by binding to an abundant intracellular chaperone protein, cyclophilin A, to form a binary complex. This complex then selectively docks onto the active, GTP-bound state of both mutant and wild-type RAS isoforms (KRAS, HRAS, and NRAS). Unlike pioneering KRASG12C inhibitors, daraxonrasib targets a broad spectrum of RAS variants common in PDAC (G12D, G12V, G12R, etc.), successfully shutting down downstream MAPK signaling regardless of the specific driver mutation or compensatory wild-type RAS activation.
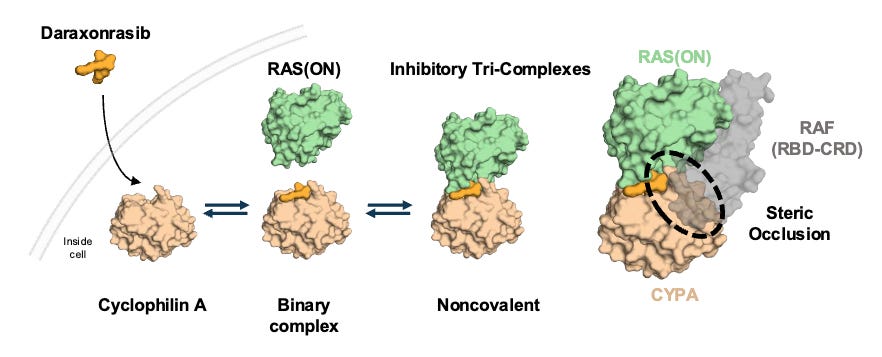
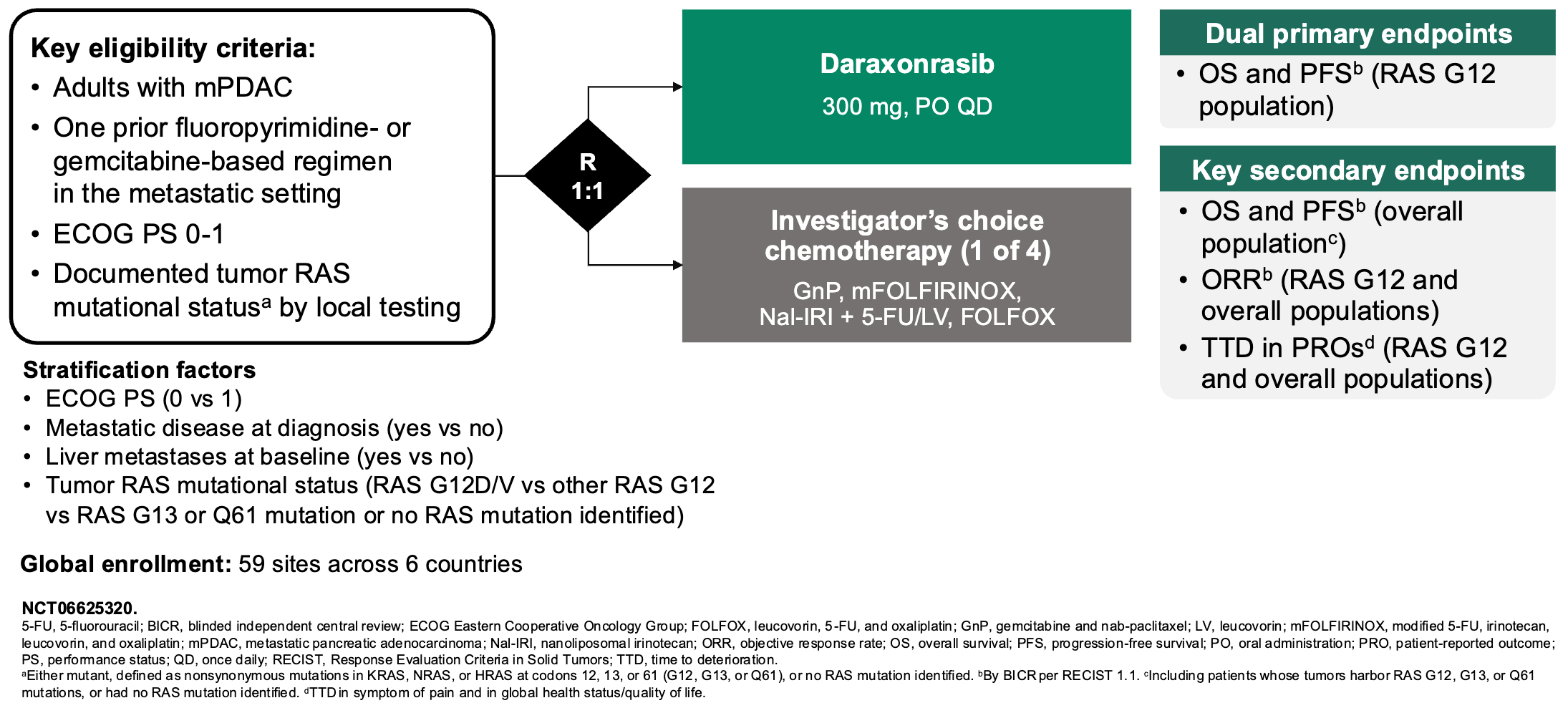
Daraxonrasib binds to intracellular cyclophilin A to form a binary complex that engages RAS(ON) and suppresses downstream signaling; Source: Revolution Medicines presentation, slide 9 Trial design: The Phase 3 RASolute 302 trial was designed to evaluate the safety and efficacy of daraxonrasib in 501 patients with metastatic pancreatic ductal adenocarcinoma (mPDAC) who had progressed on exactly one prior systemic line of therapy (typically FOLFIRINOX or Gemcitabine + nab-paclitaxel). Patients had an ECOG performance status of 0 or 1. Patients were randomized 1:1 to either daraxonrasib monotherapy administered orally at 300 mg once daily (the established RP2D) in 21-day cycles or investigator’s choice of standard-of-care (SOC) second-line chemotherapy backbones (e.g., liposomal irinotecan + 5-FU/LV, FOLFOX, or Gem/nab-paclitaxel depending on frontline exposure). The primary endpoint was Progression-Free Survival (PFS) and Overall Survival (OS) evaluated in both the overall Intent-to-Treat (ITT) population and the subset with RASG12 mutations.
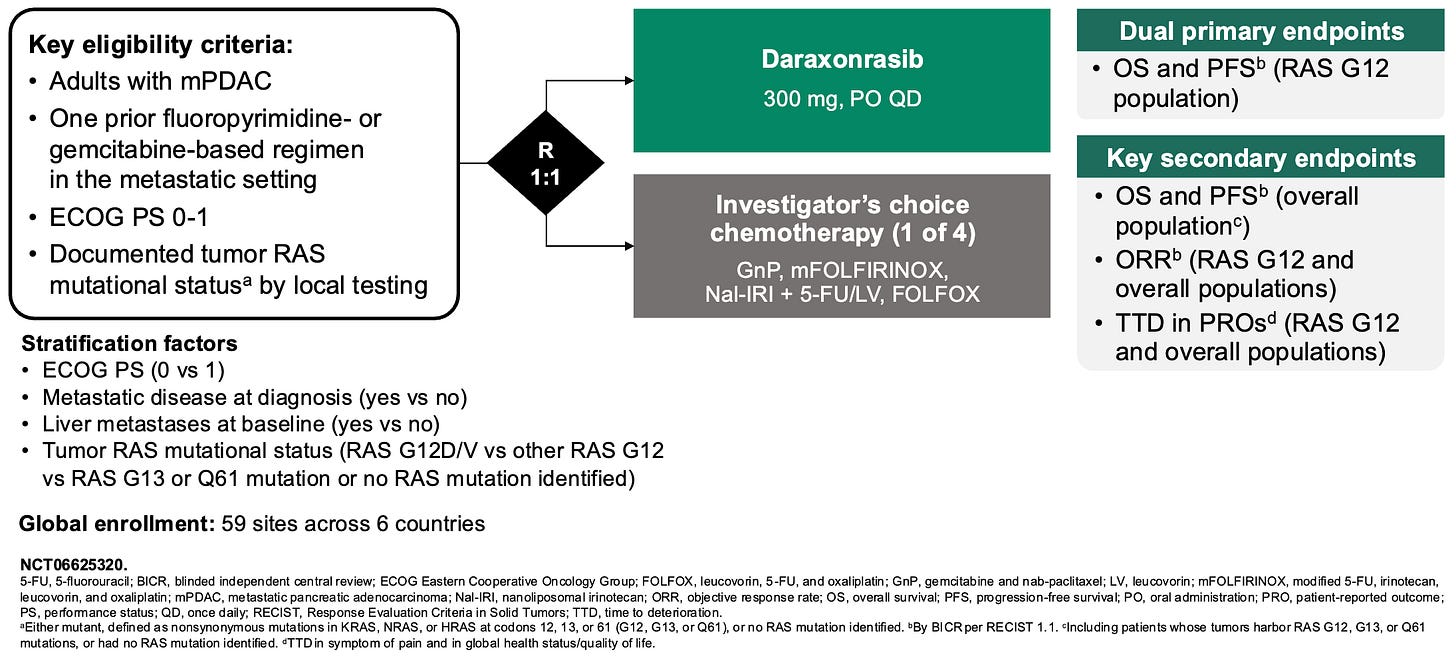
Phase 3 RASolute 302 trial design; Source: Revolution Medicines presentation, slide 9 Data (press release, slide deck, NEJM paper):
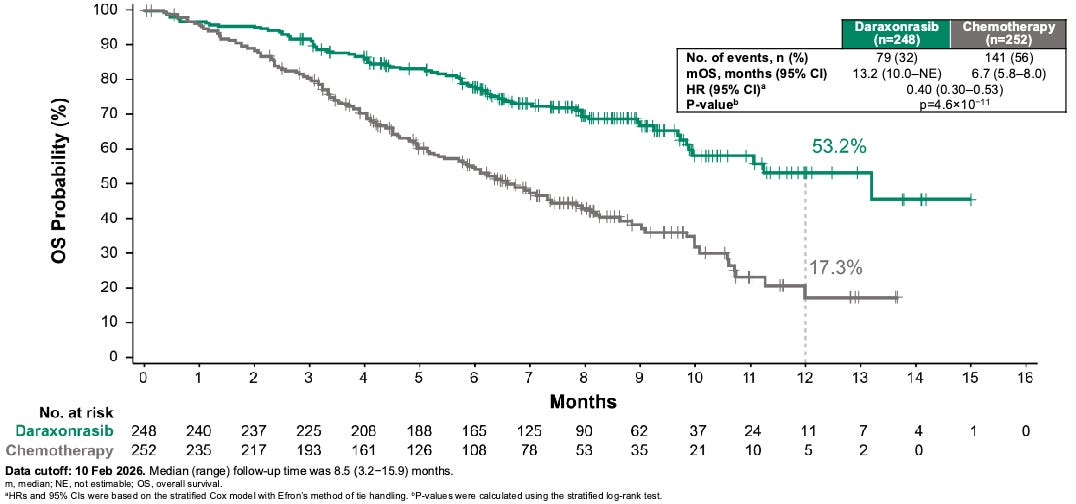
Efficacy: The trial delivered highly mature, definitive survival benefits that met both primary (mOS 13.2 months dara versus 6.7 months SOC, HR 0.4, P < 0.001; mPFS 7.2 months dara versus 3.6 months SOC) and secondary endpoints (ORR 47% dara versus ~11% SOC) with remarkable statistical significance. Furthermore, daraxonrasib delayed the median time to cancer-related symptom worsening to >9 months compared to <4 months in the chemotherapy control arm.
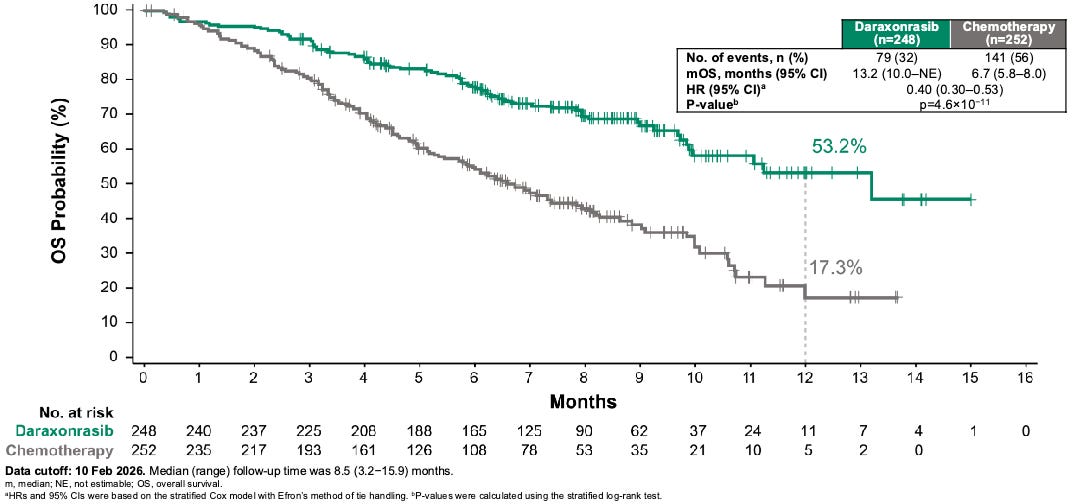
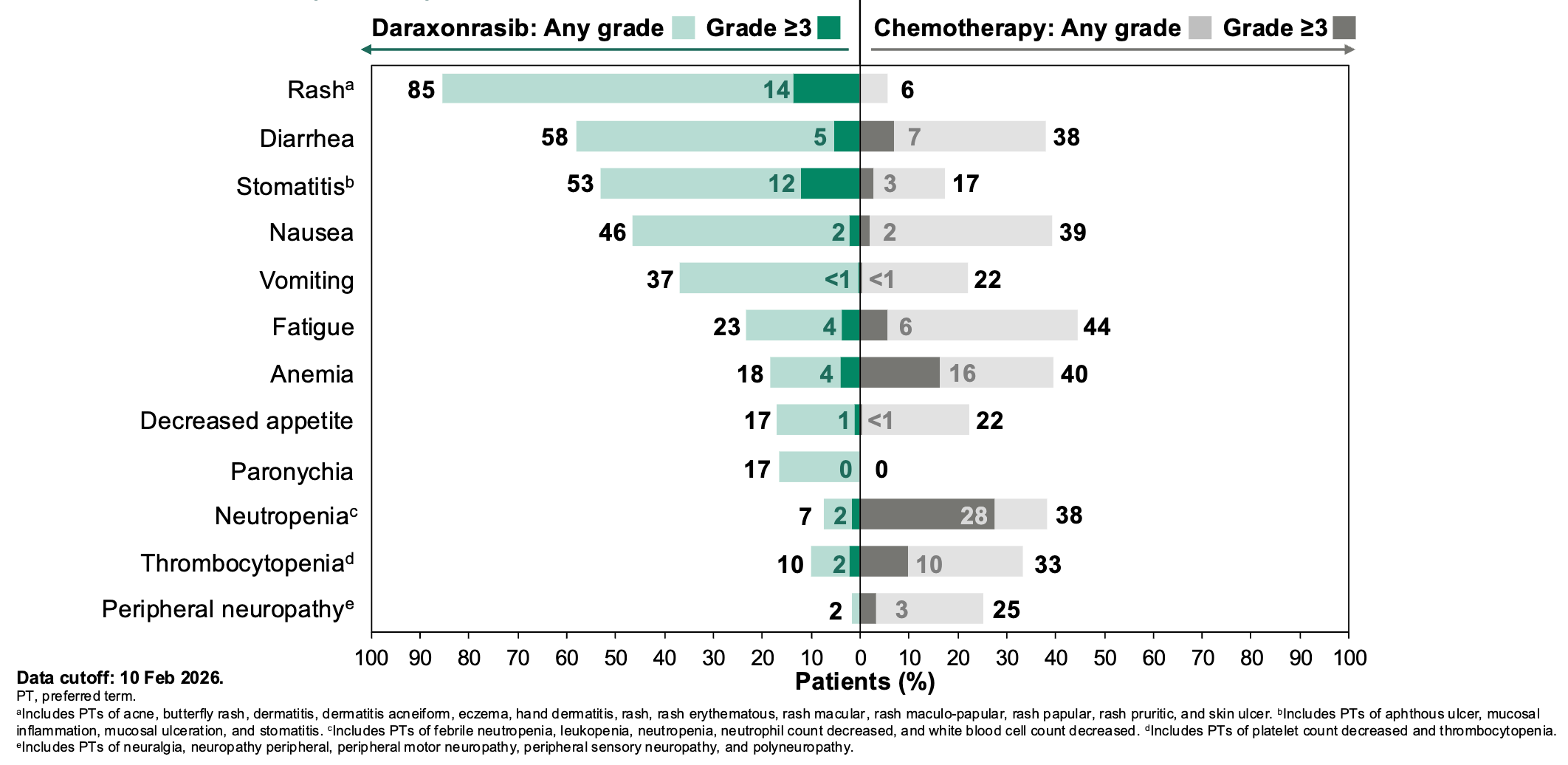
Daraxonrasib more than doubled the median overall survival (mOS) in patients with metastatic pancreatic ductal adenocarcinoma (mPDAC) from 6.7 months for standard of care to 13.2 months daraxonrasib-treated participants, resulting in a 60% relative risk reduction in death; Source: Revolution Medicines presentation, slide 14 Safety: Daraxonrasib exhibited a vastly improved therapeutic index compared to cytotoxic options. Grade ≥3 treatment-related adverse events occurred in 43.6% of the daraxonrasib cohort vs. 57.5% in the chemotherapy arm. The primary toxicities were manageable, on-target class effects: skin rash and mouth sores (stomatitis), which were easily mitigated with standard topicals or antibiotics. Crucially, the treatment discontinuation rate due to toxicities was just 1.2% for daraxonrasib compared to 11.2% for chemotherapy.
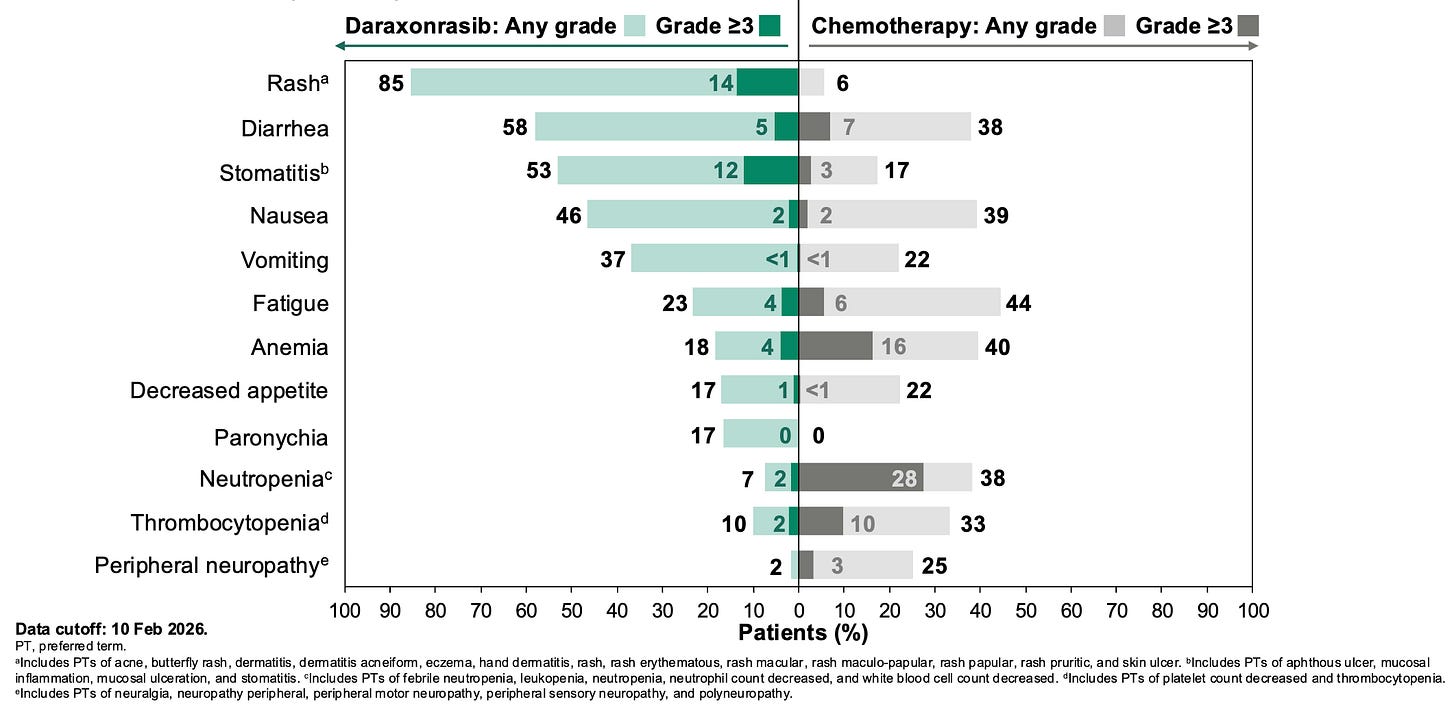
Most common (>15%) treatment-related adverse events in the Phase 3 RASolute 302 trial; Source: Revolution Medicines presentation, slide 19 Impact:
Potentially Best-in-Disease for 2L mPDAC: A 60% reduction in the risk of death (HR 0.40) and an absolute mOS extension of 6.5 months is unprecedented in second-line mPDAC, where historical increments are measured in weeks. If approved, daraxonrasib has the potential to emerge as a preferred option over standard cytotoxic single- or doublet-chemotherapy choices in the second-line setting. Shifting patients from toxic, center-based intravenous chemotherapeutic infusions to a once-daily, highly tolerable oral pill could further improve patient quality of life. This data were presented to a room full of oncologists who have seen thousands of pancreatic cancer patients die months after diagnosis, who will soon have a new tool in their tool belt; it was impressive enough to merit a standing ovation!
 Samuel Hume@DrSamuelBHumeOne of the most amazing things I’ve ever seen: a standing ovation for the full Daraxonrasib results I feel inspired and energised, to put it mildly — we have a targeted therapy for pancreatic cancer now, and nothing is undruggable anymore11:18 PM · May 31, 2026 · 1.95M Views103 Replies · 1.5K Reposts · 9.51K Likes
Samuel Hume@DrSamuelBHumeOne of the most amazing things I’ve ever seen: a standing ovation for the full Daraxonrasib results I feel inspired and energised, to put it mildly — we have a targeted therapy for pancreatic cancer now, and nothing is undruggable anymore11:18 PM · May 31, 2026 · 1.95M Views103 Replies · 1.5K Reposts · 9.51K LikesValidation of the pan-RAS Strategy: This success delivers robust clinical proof-of-concept for pan-RAS(ON) inhibition, demonstrating that wild-type sparing/multi-selectivity can achieve deep efficacy without creating unmanageable systemic toxicity.
Next steps: Having secured FDA Breakthrough Therapy and Orphan Drug designations, and being enrolled in the FDA Commissioner’s National Priority Voucher (CNPV) pilot program, a formal NDA filing is imminent. Revolution Medicines anticipates approval and commercial launch are anticipated within the next 1 to 2 months. On May 1, 2026, the FDA cleared an Expanded Access Program (EAP) protocol, allowing immediate pre-approval clinical access to the drug for eligible relapsed mPDAC patients who cannot wait for the final commercial rollout. Revolution Medicines is already actively enrolling the global, Phase 3 RASolute 303 trial. This study is evaluating daraxonrasib combined with standard chemotherapy versus chemotherapy alone as a first-line intervention for metastatic PDAC, aiming to move the molecule into treatment-naive settings. Driven by the pan-RAS profile, Phase 3 registrational studies are simultaneously underway for daraxonrasib in other major RAS-addicted indications, specifically advanced RAS-mutant non-small cell lung cancer (NSCLC) and colorectal cancer.

SILVER Celcuity - Gedatolisib (Pan-PI3K/mTOR inhibitor) / Phase 3 (HR+ HER2- mBC)
At ASCO 2026, the Phase 3 VIKTORIA-1 trial established gedatolisib as a major clinical success by cutting the risk of disease progression in half (HR=0.50) and doubling median PFS to 11.1 months, delivering an efficacy triumph.
Indication: Hormone receptor-positive, HER2-negative (HR+/HER2-) metastatic breast cancer (mBC) is fundamentally driven by luminal mammary epithelial proliferation fueled by hyperactive estrogen receptor (ER) and/or progesterone receptor (PR) transcription factors, alongside deregulated cyclin D-CDK4/6-Rb and PI3K/AKT/mTOR signaling pathways that promote uncontrolled cell cycle progression and survival. Over time, selective therapeutic pressure inevitably induces acquired resistance mechanism mutations, such as ESR1 ligand-binding domain mutations (enabling ligand-independent ER activation) or PIK3CA, AKT1, and PTEN alterations that hyperactivate downstream survival cascades. To counter this biology, the frontline standard of care (SOC) couples endocrine therapy—principally a non-steroidal aromatase inhibitor (letrozole or anastrozole)—with a CDK4/6 inhibitor (palbociclib, ribociclib, or abemaciclib), which dramatically delays disease progression. Upon subsequent progression, the second-line SOC pivots sharply based on liquid biopsy or genomic profiling: tumors harboring ESR1 mutations are prioritized for next-generation oral selective estrogen receptor degraders (SERDs) like elacestrant; those exhibiting PIK3CA, AKT1, or PTEN mutations transition to targeted inhibitor combinations (e.g., capivasertib or alpelisib plus fulvestrant, or multi-PAM agents like gedatolisib); while patients with germline BRCA1/2 mutations utilize PARP inhibitors (olaparib, talazoparib). Once endocrine-refractory states develop or visceral crisis is imminent, sequential cytotoxic chemotherapy or heavily targeted antibody-drug conjugates (ADCs) like sacituzumab govitecan or datopotamab deruxtecan (Dato-DXd) are deployed to sustain survival.
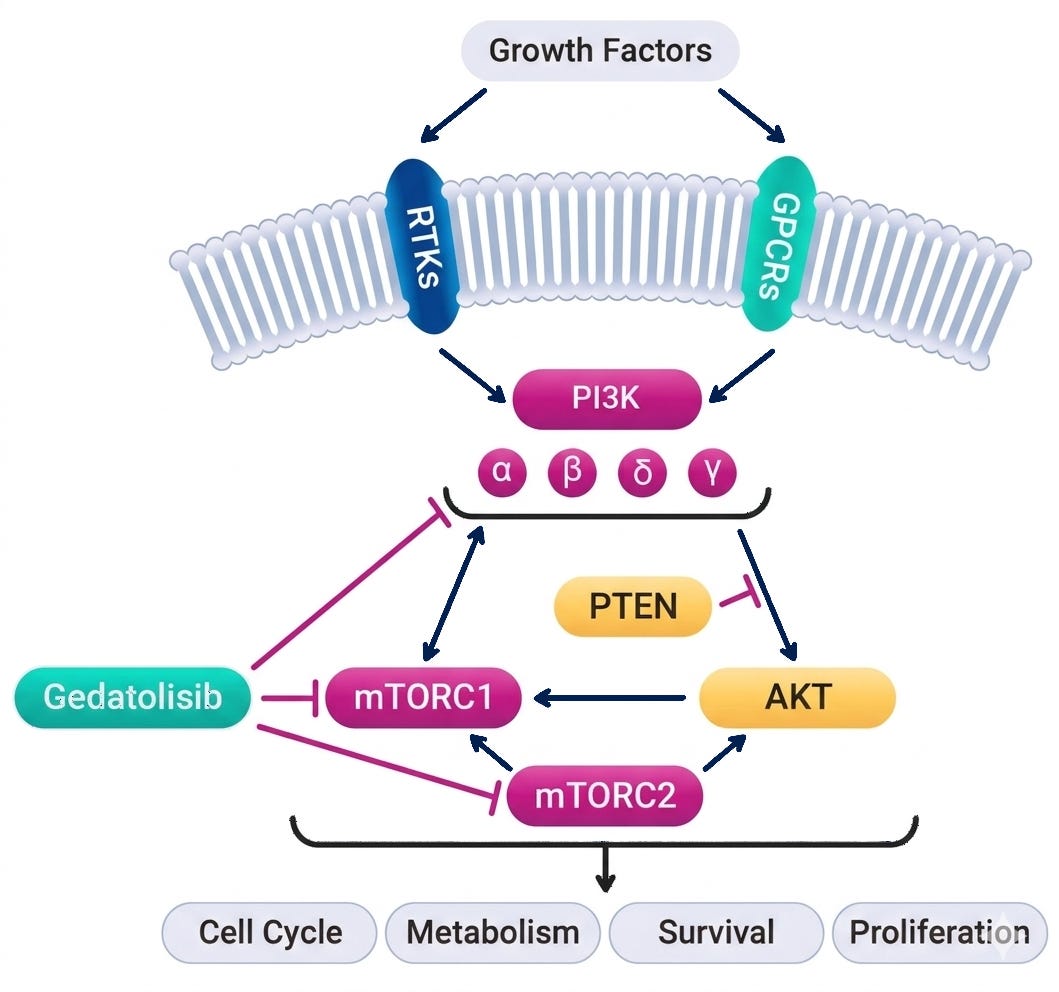
Mechanism: Gedatolisib is a highly potent, intravenously administered pan-PI3K/mTOR inhibitor designed for absolute vertical suppression of the PI3K/AKT/mTOR (PAM) pathway. Unlike single-node inhibitors (e.g., the α-selective PI3K inhibitor alpelisib), gedatolisib simultaneously targets all four class I PI3K isoforms (α, β, γ, δ) as well as both mTORC1 and mTORC2 complexes. By blocking both upstream (PI3K) and downstream (mTOR) nodes, it preempts the compensatory cross-activation and adaptive resistance loops that typically blunt the efficacy of single-target single-agent options.
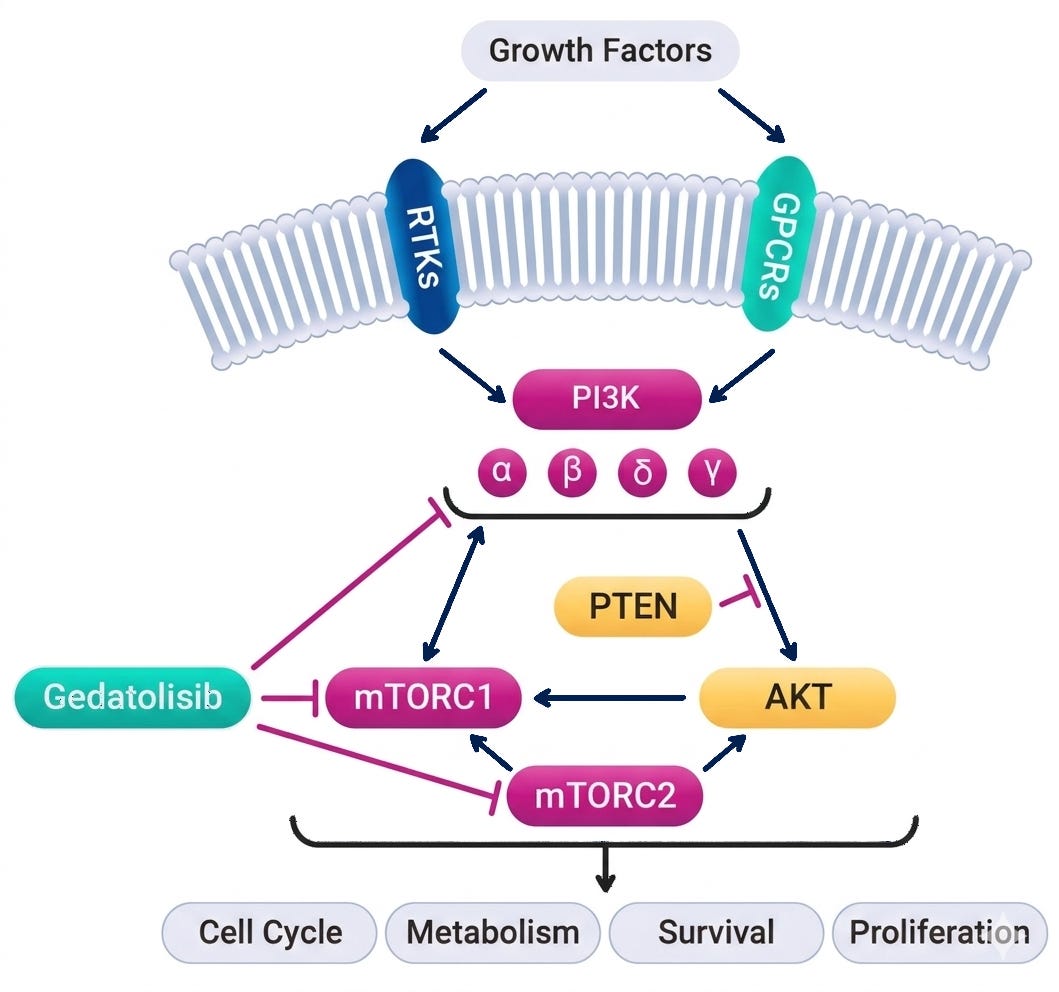
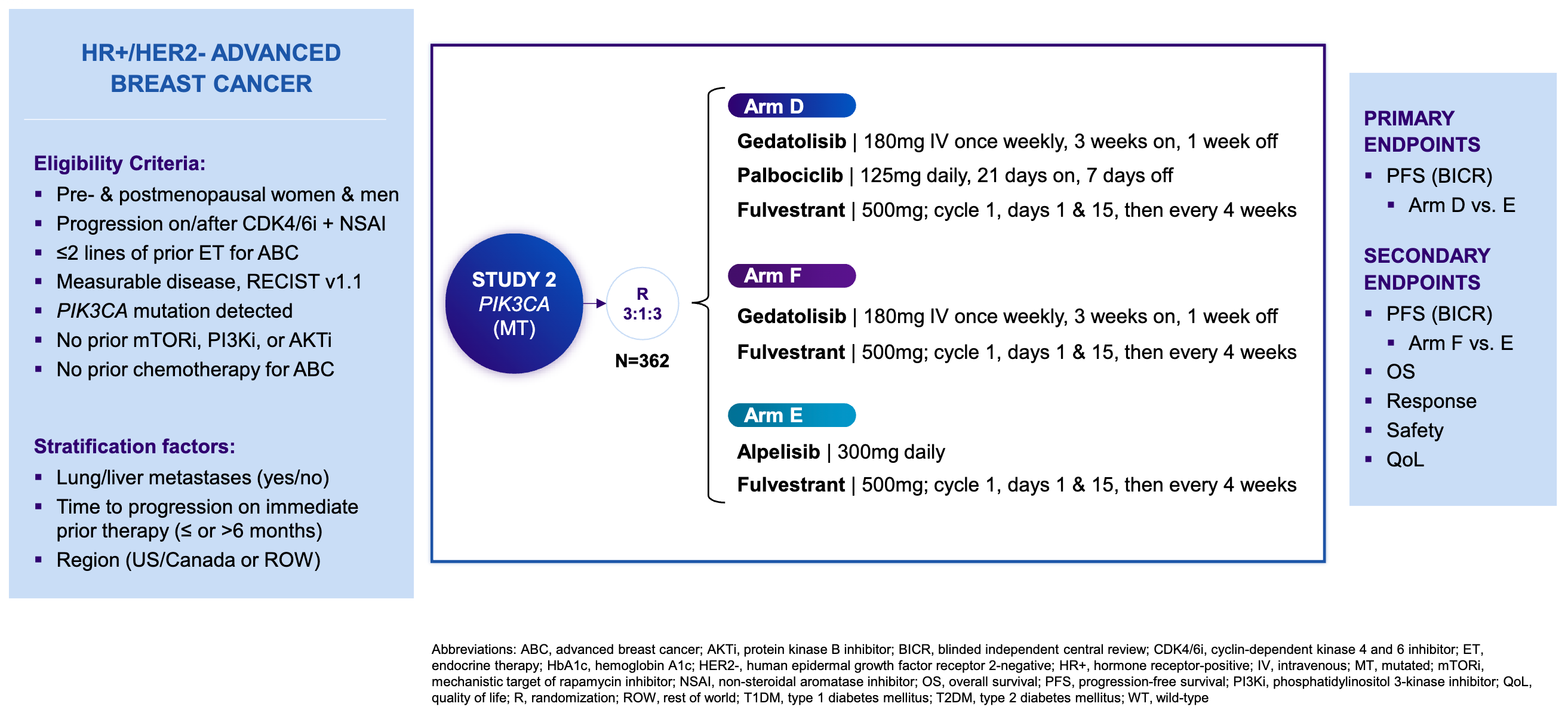
Gedatolisib mechanism of action; Source: Celcuity presentation, slide 8 Trial design: The Phase 3 VIKTORIA-1 trial was designed to evaluate the safety and efficacy of gedatolisib in 362 patients with HR+ HER2- mBC whose disease had progressed during or after standard frontline treatment with a CDK4/6 inhibitor and an aromatase inhibitor. Patients were randomized 3:3:1 to one of three treatment arms (see below). The primary endpoint was Progression-Free Survival (PFS) by Blinded Independent Central Review (BICR) for the triplet vs. active control. A key secondary endpoint evaluated the doublet vs. active control.
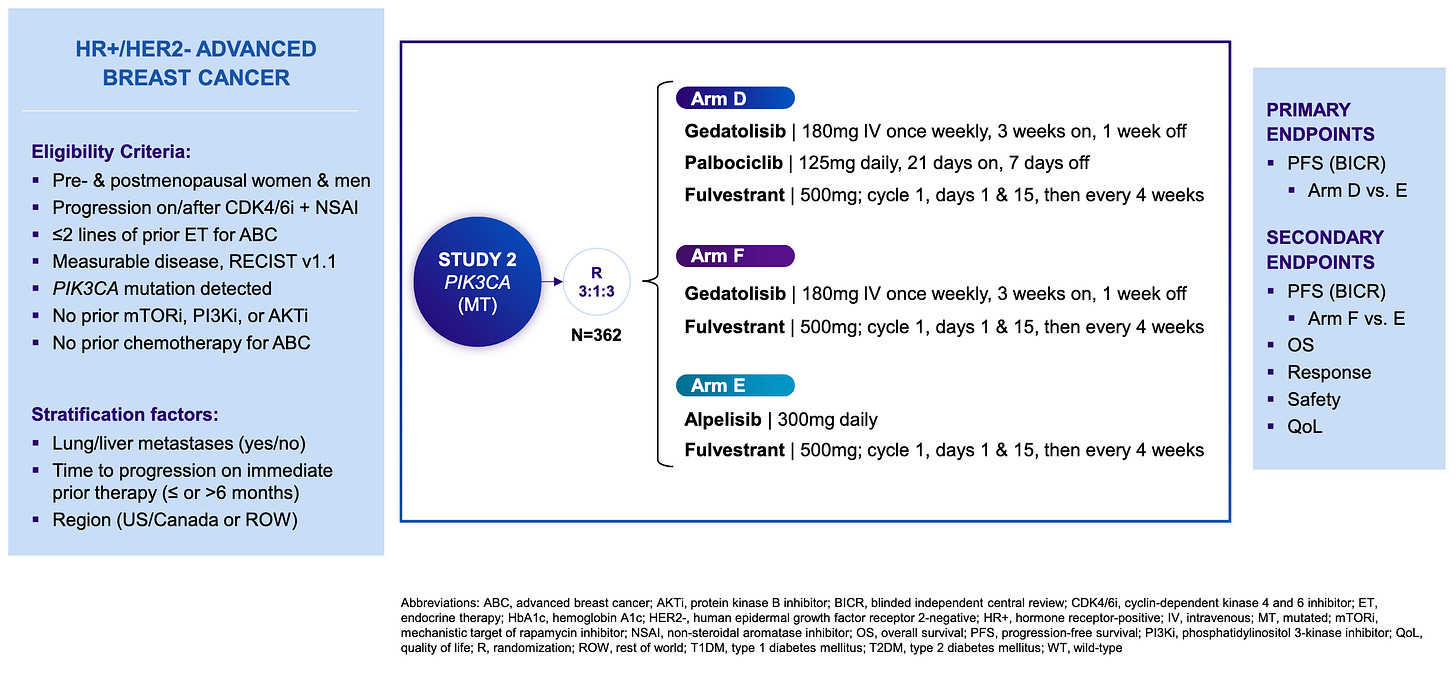
Phase 3 VIKTORIA-1 trial design; Source: Celcuity presentation, slide 9 Data (press release, slide deck):
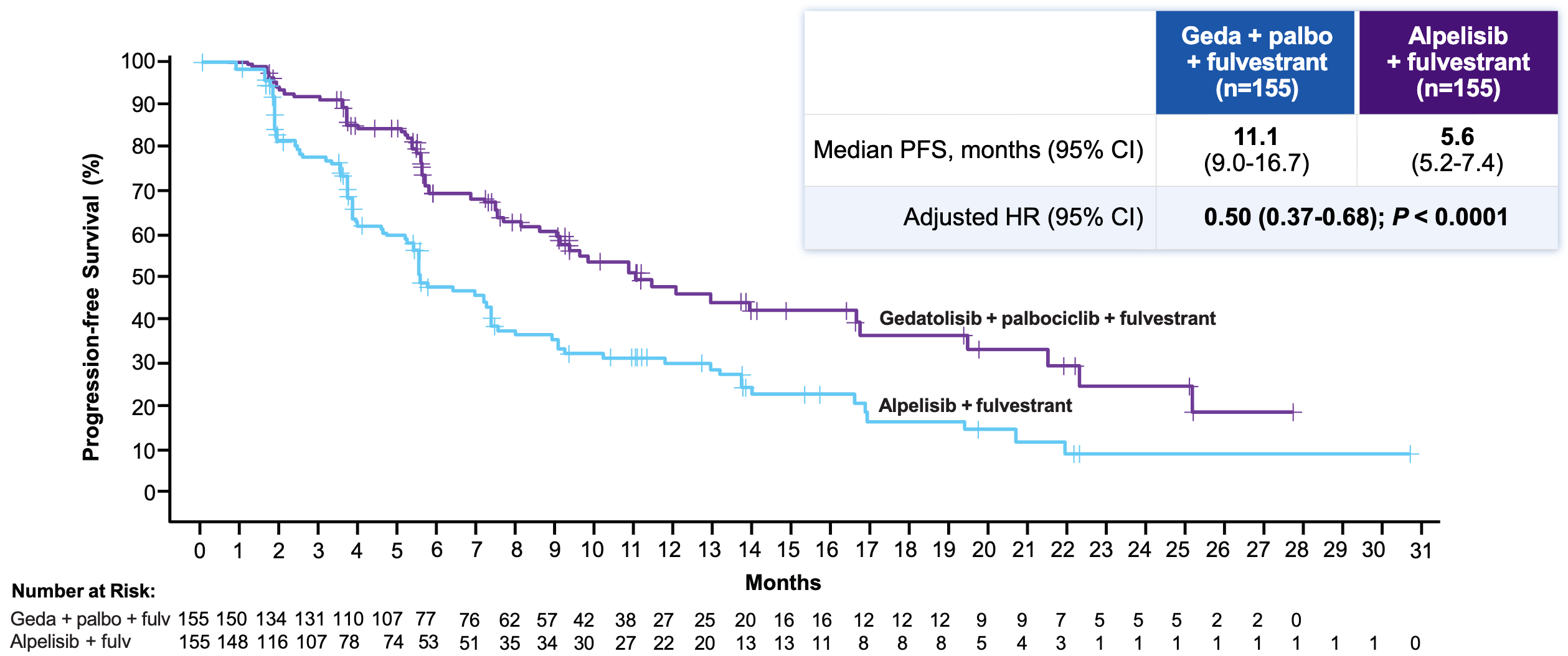
Efficacy: The trial met its primary and key secondary endpoints with high statistical significance, though the baseline numbers missed “lofty” expectations established by small, earlier phase trials (where a historical mPFS had hovered around 14.6 months).
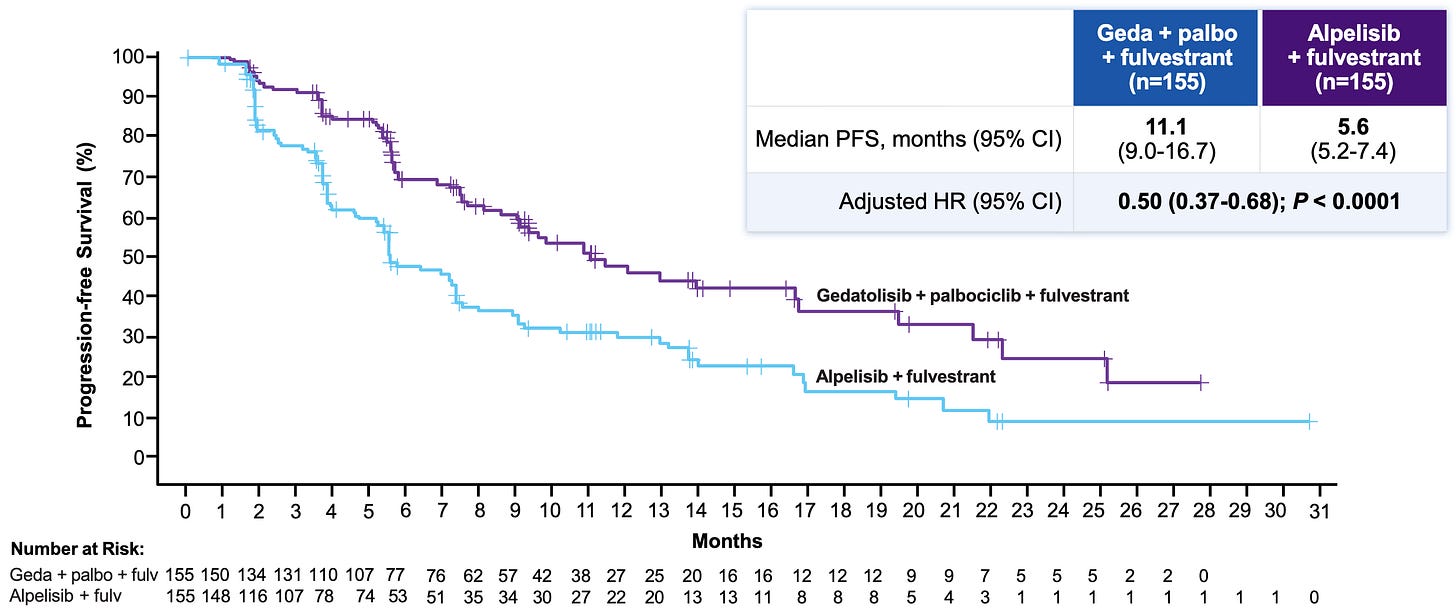
Progression-Free Survival (BICR) in the Phase 3 VIKTORIA-1 trial almost doubled from 5.6 months in the Alpelisib + Fulvestrant (Control) arm to 11.1 in the Gedatolisib Triplet arm, reducing the risk of disease progression or death by 50%; Source: Celcuity presentation, slide 13 Safety: Gedatolisib demonstrated a far superior metabolic and GI toxicity index compared to alpelisib. Grade ≥3 on-target metabolic toxicities were radically lower for gedatolisib. the gedatolisib arms saw markedly lower rates of hyperglycemia (11.5-15% geda versus 57.9% alpelisib), diarrhea (9.6-15% geda versus 40.1% alpelisib), and discontinuation Rates due to treatment-related adverse events (2.6-3.8% geda versus 7.1% alpelisib). The trade-off toxicity for gedatolisib was stomatitis (mouth inflammation), appearing in ~61% of patients across both gedatolisib arms versus 34.2% in the alpelisib arm.
Impact:
Efficacy Nearly Doubling over Standard of Care: Clinically, cutting the risk of disease progression or death by half (HR = 0.50) and doubling the mPFS from 5.6 to over 11 months represents a massive win for a second-line, post-CDK4/6 inhibitor mutant population.
Potentially Broad Label: Since gedatolisib is equally potent in both PIK3CA-mutant and PIK3CA-wild-type contexts, it could avoid the niche labeling restrictions of single-node competitors, pending regulatory review.
Next steps: The FDA is currently reviewing its NDA for the PIK3CA wild-type (WT) advanced breast cancer cohort (based on prior VIKTORIA-1 data that showed a 7.3-month PFS advantage over fulvestrant alone). The assigned PDUFA goal date is July 17, 2026. Celcuity has confirmed plans to submit this newly presented PIK3CA-mutant data package to the FDA in an sNDA during Q3 2026 to expand the commercial label broadly across both mutational categories. In the clinic, oncologists will need to dissect the overlapping utility of the triplet vs. the doublet. Given the nearly identical PFS curves and the cleaner hematologic safety profile of the doublet, real-world positioning may favor dropping the CDK4/6 backbone in patients sensitive to cytopenias, while utilizing the triplet for highly aggressive kinetics where maximizing the initial ORR (48.9%) is paramount.
BRONZE Ideaya - Darovasertib (PKC inhibitor) / Phase 2/3 (MUM)
At ASCO 2026, the late-breaking oral presentation of the randomized Phase 2/3 OptimUM-02 trial (Abstract LBA9503) positioned IDEAYA Biosciences and Servier’s darovasertib + crizotinib combination as the definitive new frontline standard of care for HLA-A*02:01-negative metastatic uveal melanoma (mUM), a historically recalcitrant population with zero approved targeted or systemic options.
Indication: HLA-A*02:01-negative metastatic uveal melanoma (mUM) is a molecularly distinct ocular malignancy predominantly driven by early, mutually exclusive, activating mutations in the G-protein α subunit genes GNAQ or GNA11 (occurring in over 90% of cases). These driver mutations constitutively hyperactivate the downstream Protein Kinase C (PKC) and MAPK/MEK pathways, fueling rapid cellular proliferation and a profound tropism for hepatic metastasis, where the tumor manipulates a dense, immunosuppressive microenvironment characterized by low tumor mutational burden (TMB) and poor T-cell infiltration. Because of this non-inflamed “cold” tumor phenotype, standard cutaneous melanoma treatments like single-agent anti-PD-1 blockades are largely ineffective. For the roughly 55% of patients who test HLA-A*02:01-negative, the bispecific gp100-directed T-cell engager tebentafusp (Kimmtrak) is biologically restricted and unavailable. Consequently, the historical standard of care has defaulted to combination immune checkpoint inhibition with ipilimumab plus nivolumab or enrollment in clinical trials utilizing liver-directed therapies (such as hepatic artery infusion or percutaneous hepatic perfusion), though response rates to standard immunotherapy remain dismally low (~5%) and prognoses poor. This therapeutic void has positioned newly validated molecular targeted strategies, such as dual vertical inhibition of the PKC and c-Met pathways, as the definitive frontline standard to directly intercept the underlying GNAQ/11 biology in this underserved population.
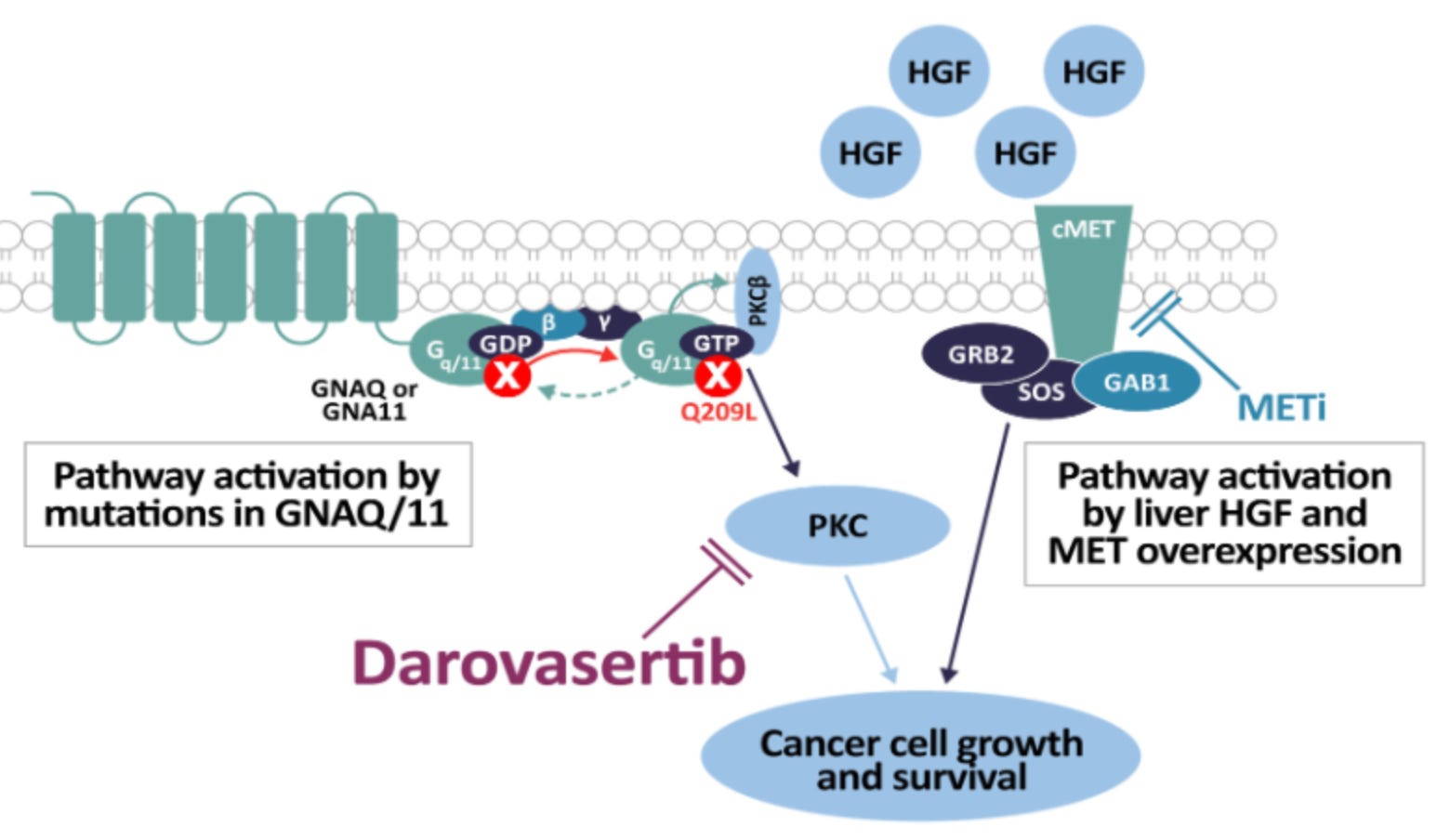
Mechanism: Darovasertib is a first-in-class, oral small-molecule Protein Kinase C (PKC) inhibitor. Up to 95% of uveal melanomas harbor activating mutations in the G-protein subunits GNAQ or GNA11. This oncogenic mutation continuously hyperactivates the downstream PKC signaling pathway, fueling tumor proliferation. While single-agent PKC inhibition faces feedback resistance, combining it with crizotinib (Xalkori), an oral c-Met/MET inhibitor—blocks parallel pathway activation triggered by hepatic growth factor (HGF) and MET overexpression. Because mUM heavily metastasizes to the liver (a high-HGF environment), this dual vertical inhibition significantly enhances synthetic lethality and prevents tumor escape.
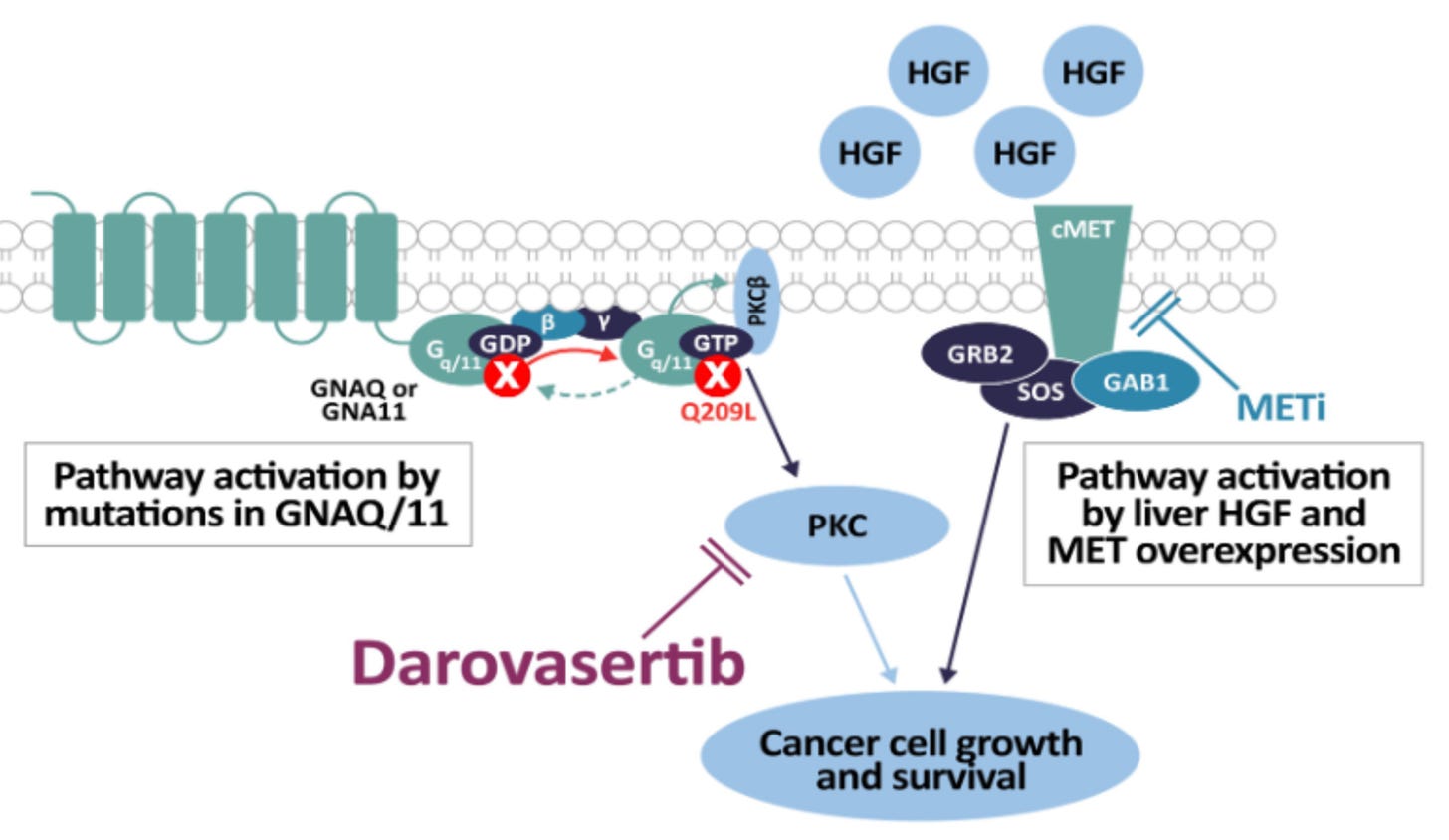
Darovasertib mechanisms of action; Source: Ideaya presentation, slide 2 Trial design: The Phase 2/3 OptimUM-02 trial was designed to evaluate the safety and efficacy of darovasertib in 313 patients with treatment-naive (first-line) patients with histologically or cytologically confirmed metastatic uveal melanoma who tested HLA-A*02:01 negative. Patients were randomized to either darovasertib (300 mg BID) + crizotinib (200 mg BID) administered orally in continuous cycles or investigator’s Choice of Therapy reflective of current real-world checkpoint inhibitor use (~77% received the doublet ipilimumab + nivolumab; others received pembrolizumab or dacarbazine). The primary endpoint of the Phase II portion was Progression-Free Survival (PFS) by Blinded Independent Central Review (BICR). The dual primary endpoint of the ongoing Phase 3 portion is Overall Survival (OS).
Data (press release, slide deck):
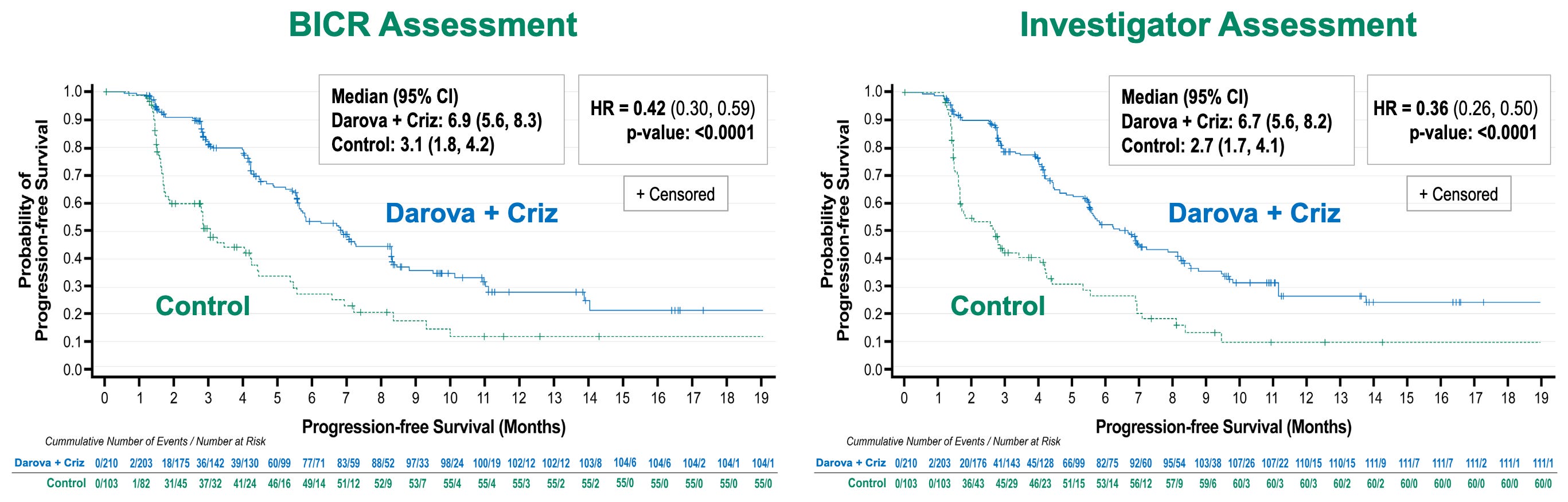
Efficacy: The ASCO presentation reported a clinically meaningful and statistically significant improvement in PFS with darova + criz vs control, with a 58-64% reduction in the risk of disease progression or death (p < 0.0001) for BICR and investigator assessments (see below). OS data are not yet mature, but a pre-specified interim look demonstrated an early trend favoring the darovasertib combination.
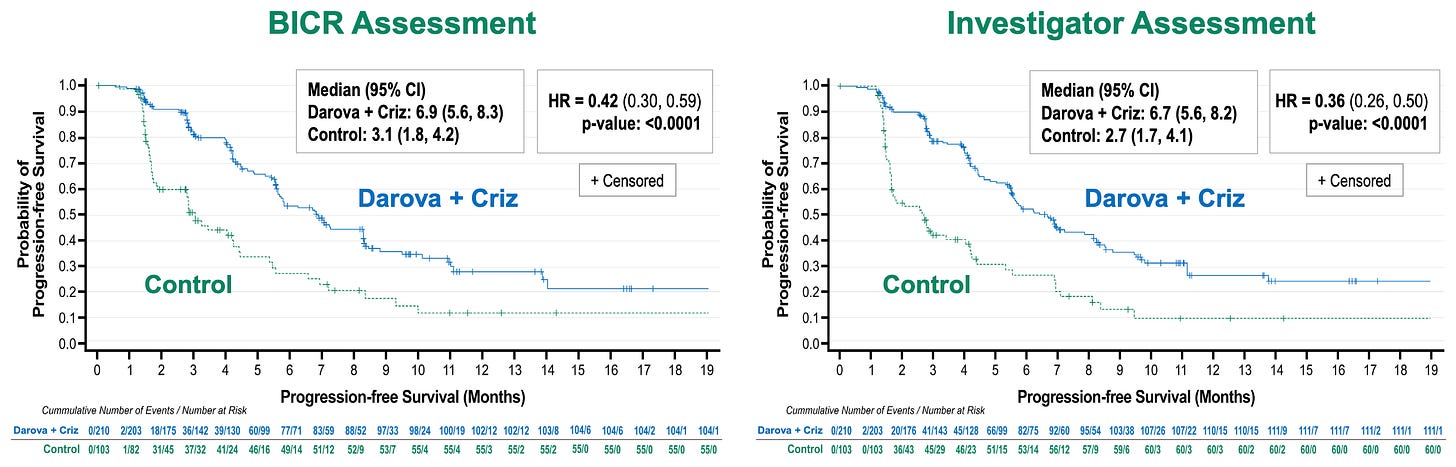
Primary efficacy endpoint results in Phase 2/3 OptimUM-02 trial; Source: Ideaya presentation, slide 8 Safety: The oral targeted combination proved remarkably manageable and safer than toxic checkpoint doublets. Grade 3/4 TRAEs were comparable between arms (40.6% combo vs. 37.0% ICT). The most common Grade 3+ events for the combo were class-specific: diarrhea, syncope, and hypotension. Treatment-Related Serious AEs (TR-SAEs) were radically lower for the targeted combo (9.2%) versus the checkpoint control arm (25.0%). Discontinuations due to TRAEs were exceptionally low for darovasertib at 2.5% (10% for crizotinib) compared to 19.0% in the ICT immunotherapy backbone.
Impact:
Resolving a Massive Genomic Blindspot: Immunotherapies like tebentafusp (Kimmtrak) changed the game in uveal melanoma but are strictly restricted to HLA-A*02:01 positive patients (~45% of the population). OptimUM-02 establishes an effective option for the remaining ~55% who are HLA-A*02:01 negative and otherwise faced poor prognoses on standard cutaneous melanoma regimens.
Defeating the Immunotherapy Paradigm: Proving a targeted oral doublet can achieve an HR of 0.42 and a 37.1% response rate against a backdrop where ~77% of patients received ipilimumab/nivolumab establishes molecular targeting as vastly superior to immune checkpoint blockade in GNAQ/11-driven ocular malignancies.
Next steps: In late April 2026, the FDA accepted darovasertib + crizotinib into the Oncology Center of Excellence Real-Time Oncology Review (RTOR) program. IDEAYA has already initiated the rolling submission process. They expect to complete the formal New Drug Application (NDA) filing in H2 2026, paving the way for an expedited approval pathway. Clinically, because early phase data showed darovasertib also works well regardless of HLA status, IDEAYA is exploring expansion strategies into HLA-A*02:01 positive mUM. More importantly, active registrational studies are underway testing darovasertib in the neoadjuvant and adjuvant settings for localized primary uveal melanoma to prevent metastatic recurrence entirely.
Honorable Mention Kelonia / Eli Lilly - KLN-1010 (in vivo BCMA CAR-T) / Phase 1/2 (MM)
The clinical data presented for KLN-1010 at ASCO 2026 backs up Eli Lilly’s massive $7 billion acquisition of Kelonia Therapeutics. By generating CAR-T cells directly inside the patient’s body, the candidate bypasses the severe toxicities, delays, and manufacturing hurdles that limit traditional cellular therapies.
Indication: Multiple myeloma is a hematologic malignancy characterized by the clonal proliferation of malignant plasma cells within the bone marrow microenvironment. Pathophysiologically, this expansion is driven by genetic abnormalities (such as t(11;14) translocations or del(17p)) and hyperactive signaling pathways (including NF-κB and JAK/STAT), which trigger the overproduction of a monoclonal immunoglobulin (M-protein) and disrupt normal hematopoiesis and bone remodeling. This results in the classic CRAB clinical features: Calcium elevation, Renal insufficiency, Anemia, and osteolytic Bone lesions. The contemporary frontline standard of care relies heavily on a quadruplet induction regimen combining an anti-CD38 monoclonal antibody (e.g., daratumumab or isatuximab), a proteasome inhibitor (e.g., bortezomib), an immunomodulatory drug (e.g., lenalidomide), and dexamethasone. For transplant-eligible patients, this intensive induction is followed by high-dose chemotherapy conditioning and an autologous stem cell transplant (ASCT), followed by maintenance therapy designed to sustain minimal residual disease (MRD) negativity. When the disease inevitably enters the relapsed/refractory (RRMM) setting, the therapeutic paradigm shifts toward highly active, novel immunotherapies, specifically BCMA-targeted CAR-T cell therapies (such as cilta-cel) and T-cell engaging bispecific antibodies (such as teclistamab or talquetamab).
Mechanism: KLN-1010 is an off-the-shelf, intravenously administered in vivo anti-BCMA CAR-T cell therapy. It utilizes Kelonia’s proprietary in vivo Gene Placement System (iGPS), which leverages a highly optimized, replication-incompetent lentiviral vector. The vector particles feature precise envelope modifications and targeting molecules that specifically home in on and transduce endogenous T cells inside the body, steering clear of non-target cells. Once the vector docks, it delivers the genetic cargo to integrate and express the anti-BCMA CAR molecule directly on the patient’s circulating T cells, transforming them into tumor-killing effectors in situ.
Trial design: Phase I inMMyCAR open-label, multi-center, dose-escalation study (NCT07075185) tested KLN-1010 in 18 heavily pretreated, highly refractory patients with Relapsed/Refractory Multiple Myeloma (MM). Unlike commercial ex vivo CAR-Ts that require harsh fludarabine/cyclophosphamide chemotherapy to clear space in the bone marrow, KLN-1010 was infused via a single IV line into non-chemically depleted patients.
Data (press release):
Efficacy: The interim results drew immense praise at the ASCO meeting. 100% (18/18 patients) responded to the single infusion and achieved minimal residual disease (MRD)-negative bone marrow status at day 30 post-infusion. For the cohort with ≥4 months of extended follow-up (n=6), responses continued to deepen over time. 4 patients achieved a Stringent Complete Response (sCR) and 2 achieved a Very Good Partial Response (VGPR). The earliest dosed patients show ongoing, deep MRD-negative persistence beyond 9 to 10 months. Correlative tracking confirmed that in vivo CAR-T kinetics and persistence in the peripheral blood and bone marrow exceeded the traditional curves frequently seen in standard ex vivo manufacturing, even across diverse baseline T-cell phenotypes.
Safety: While 16 out of 18 patients experienced CRS, it was notably mild and transient, entirely restricted to Grade 1 or 2. No Grade ≥3 CRS occurred. There were 2 cases of early-onset neurotoxicity: 1 Grade 1 and 1 Grade 3. Both resolved completely with standard management. Crucially, there were zero cases of delayed-onset neurotoxicity (such as Parkinsonian symptoms) and no severe, prolonged cytopenias.
Impact:
True Outpatient Democratization: Since it cuts out lymphodepletion and keeps severe CRS at zero, the Safety Review Committee officially backed outpatient dosing. This shifts CAR-T away from specialized academic ICUs and into community oncology centers.
Erasing the Vein-to-Vein Wait: Standard cell manufacturing takes 4 to 8 weeks, a window in which aggressive myeloma often progresses. KLN-1010 registered a median time of just 13 days from patient consent to infusion, essentially an off-the-shelf biologic timeline.
Validation of Lilly’s $7B Bet: Eli Lilly executed the acquisition of Kelonia for $3.25 billion upfront (escalating to $7 billion with milestones) just weeks before ASCO. Lilly executives publicly labeled the 100% response rate as “nutty,” solidifying their immediate leadership in the next wave of genetic medicine alongside competitor AstraZeneca (whose rival in vivo lentiviral asset, ESO-T01, did not present new data at this meeting).
Next steps: Kelonia (now operating under Lilly) is concluding its dose-escalation cohorts to formally establish the Recommended Phase 2 Dose (RP2D) and open an expanded Phase 2 efficacy cohort under its newly granted FDA Fast Track designation. Backed by the clean safety profile and lack of cytopenias, clinical strategies are already being drawn up to move KLN-1010 into frontline or early-relapse multiple myeloma, where a patient’s healthier T-cell fitness could yield even more durable in vivo kinetics. Lilly plans to leverage the underlying iGPS lentiviral vector platform to swap out the BCMA binder for CD19/CD20 or solid tumor targets, scaling the asset across a wider oncologic pipeline.
To contact us, please send us an email at biotechreadout@gmail.com
Disclaimers
Investigational Status Disclaimer
The therapeutic candidates discussed in this newsletter are currently in clinical development and have not been approved for commercial sale by the U.S. Food and Drug Administration (FDA), the European Medicines Agency (EMA), or other global regulatory authorities. Their safety and efficacy have not been established. References to pipeline products and ongoing clinical trials involve significant risks and uncertainties. Statements regarding the potential safety, potency, or efficacy of investigational drugs reflect current hypotheses and are not a guarantee of future performance or regulatory clearance. The outcome of clinical trials is inherently unpredictable, and clinical results from earlier stages may not be predictive of results in later, larger-scale trials.
No Medical Advice Disclaimer
This newsletter is for informational and educational purposes only. The content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or another qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read in this publication.
No Patient-Provider Relationship Disclaimer
The information provided in this newsletter is for educational and analytical purposes only. Receipt of this information, or any interaction with this content, does not create a physician-patient, pharmacist-patient, or any other professional-provider relationship between you and the authors or publishers. This newsletter should not be used as a substitute for a personal consultation with a qualified healthcare professional.
Forward-Looking Statements Disclaimer
This newsletter contains “forward-looking statements” regarding future events, including clinical trial timing, regulatory milestones, and projected market performance. These statements are based on current expectations and assumptions that are subject to significant risks and uncertainties. Actual results may differ materially from those expressed or implied. We undertake no obligation to update these statements as a result of new information or future developments.
Third-Party Links & Content Disclaimer
This newsletter contains links to third-party websites, including clinical trial registries and corporate presentations. Biotech Readout does not endorse, guarantee, or assume responsibility for the accuracy or reliability of any information offered by third-party providers.
Errors and Omissions Disclaimer
While we strive for technical accuracy, the information in this newsletter is provided on an “as is” basis with no guarantees of completeness, accuracy, or timeliness. Biotech Readout assumes no liability for any errors or omissions in the content of this publication.
Non-Endorsement Disclaimer
Any reference to specific commercial products, processes, or services by trade name, trademark, or manufacturer does not constitute or imply an endorsement or recommendation by the author. All trademarks are the property of their respective owners.
No Investment Advice Disclaimer
This newsletter is for informational purposes only and does not constitute financial, investment, or legal advice. The author is not a registered investment advisor. You should consult with a professional financial advisor before making any investment decisions. The biotechnology sector is highly volatile; past performance is not indicative of future results.
Conflict of Interest Disclaimer
The author of this newsletter maintains a position of independence. At the time of publication, the author holds no direct financial interest, equity, or options in any of the companies mentioned in this report. No compensation has been received from any third party to feature or analyze specific therapeutic candidates or corporate entities.