
Approvals for cholesterol-lowering and breast cancer, another tau attempt in Alzheimer's, and more
Weekly Readout #14: Week ending July 17, 2026

Disclaimer: This newsletter is for educational and informational purposes only and does not constitute medical, investment, or financial advice, nor does it establish a provider-patient relationship. Content may include forward-looking statements and discussions of investigational therapeutic candidates that are not FDA/EMA approved; their safety and efficacy remain unestablished and clinical outcomes are unpredictable. While we strive for accuracy, all information is provided as is without guarantees. As of the date of publication, the author holds no direct equity positions in the specific companies mentioned in this issue nor receives third-party compensation for this coverage. Please find a complete version of our disclaimers at the bottom of this article and on our About page.

Press ⬇️ to go down to a section and ⬆️ to go back up to the Table of Contents
Table of Contents
This week, we discuss:
China Biotech
Acquisitions ⬇️
Eli Lilly to Acquire AtaiBeckley
Approvals
Merck - Lipfendra (oral PCSK9 inhibitor) / Approved (hypercholesterolemia & HeFH) ⬇️
Celcuity - Revtorpyk (Pan-PI3K/mTOR inhibitor) / Approved (2L HR+ HER2- BC) ⬇️
Biogen - Leqembi IQKLIK (anti-Aβ mAb) / Approved (Alzheimer’s) ⬇️
Pfizer - Padcev (Nectin-4 ADC) / Approved (MIBC) ⬇️
Novartis - Fabhalta (factor B inhibitor) / Approved (IgAN) ⬇️
Clinical Trial Data
Q32 Bio - Bempikibart (anti-IL-7Rα mAb) / Phase 2a (alopecia) ⬇️
Veradermics - VDPHL01 (ER oral minoxidil) / Phase 2 (female androgenetic alopecia) ⬇️
Biogen - Diranersen (tau ASO) / Phase 2 (Alzheimer’s) ⬇️
Ipsen - Iqirvo (PPARa/d dual agonist) / Phase 3b (PBC) ⬇️
Takeda - Zasocitinib (TYK2 inhibitor) / Phase 3 (psoriasis) ⬇️
Merck - Sac-TMT (TROP2 ADC) / Phase 3 (1L PDL1-negative metastatic non-sq NSCLC) ⬇️
GSK - Jemperli (anti-PD1 mAb) / Phase 3 (Stage 3/4 MMRd rectal cancer) ⬇️
China Biotech
Financing Activity for China versus Rest of World
for the week ending July 17, 2026
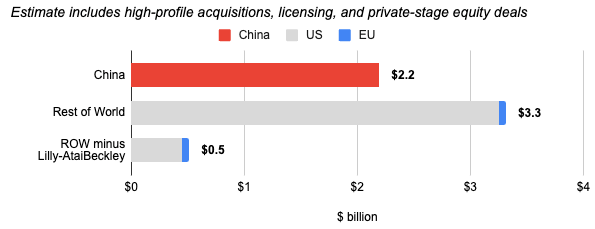
U.S Biotech Startups Have Become More Secretive to Protect Their IP
This week, both WSJ and Endpoints reported that U.S. biotech startups are adopting unprecedented levels of secrecy, using code names in offices and withholding crucial details (such as specific genes or protein targets) from websites, conferences, and even their own investors. This marks a dramatic shift for an industry that historically relied on open scientific collaboration and public disclosures after funding milestones. According to numerous industry executives and investors, this paranoia is driven by the rapid evolution, scale, and agility of the Chinese biopharma industry. Chinese companies have mastered the ability to quickly optimize, scale, and reverse-engineer experimental therapies originating in Western labs. Backed by cheaper lab staff, reagents, and clinical development pipelines, Chinese firms can fast-follow and outpace US drugmakers the moment a drug target or patent becomes public. The shift toward genetic and RNA-based therapies (such as siRNA and mRNA) has amplified these fears. Since drug targets and genes cannot be patented (only the specific drug structure can), identifying the target biology is where most of a company’s value lies. For RNA therapies, knowing the target sequence makes the drug incredibly easy to copy, forcing leading companies to hide their pipelines until they are ready for clinical trials. To defend their intellectual property, US biotechs are altering their behaviors:
Delayed Patenting: Companies are delaying patent filings or repeatedly pulling and refiling provisional patents to avoid public disclosure.
Calls for Reform: Industry leaders are advocating for patent reforms, such as extending the timeline before a patent is published or keeping certain filings redacted/classified.
Heightened Espionage Fears: Trade organizations like the Biotechnology Innovation Organization (BIO) have warned members to be wary of potential investors or partners who may actually be gathering intelligence for industrial espionage.
While some experts note that political tensions may lead to scapegoating China and that China’s internal IP protection has actually improved, the overarching trend is clear. Industry leaders warn that this pervasive secrecy in response to increasing competition from China is creating a chill in the scientific community, drastically reducing the communication and collaboration that historically drove major medical breakthroughs.
Sources: Endpoints article, WSJ article

Acquisitions

Approvals
Merck - Lipfendra (oral PCSK9 inhibitor) / Approved (hypercholesterolemia & HeFH)
On July 16, 2026, the U.S. Food and Drug Administration (FDA) approved Lipfendra (enlicitide), developed by Merck, as the first and only once-daily oral PCSK9 inhibitor. Lipfendra is indicated as an adjunct to diet and exercise to reduce low-density lipoprotein cholesterol (LDL-C) in adults with hypercholesterolemia or heterozygous familial hypercholesterolemia (HeFH), an inherited genetic disorder that limits the body’s ability to clear LDL-C from the bloodstream. It represents a major shift in the second line treatment of high cholesterol (patients who are uncontrolled with statins), as previous FDA-approved PCSK9 inhibitors were exclusively available as injections. Lipfendra uses a novel macrocyclic peptide technology that allows it to be taken as a 20 mg oral tablet. The tablet must be taken once daily in the morning, ideally 30 minutes before consuming food. Merck has indicated plans to offer patient coupons and make the drug available through the TrumpRx direct-to-consumer sales channel.
The approval was supported by two pivotal Phase 3 trials from the CORALreef clinical program. CORALreef Lipids in patients with hypercholesterolemia demonstrated a 56% reduction in LDL-C compared to placebo at week 24 and CORALreef HeFH demonstrated a 59% reduction in LDL-C compared to placebo at week 24. While Lipfendra is proven to lower LDL-C, researchers are currently conducting the CORALreef Outcomes trial (involving over 14,500 participants) to determine if this reduction translates into a decrease in major adverse cardiovascular events (MACE), which includes heart attacks, strokes, cardiovascular-related death and sometimes hospitalization for unstable angina or coronary revascularization. In clinical trials, the safety profile of Lipfendra was generally similar to that of a placebo. In the HeFH trial, the most commonly reported adverse reactions occurring more frequently than with placebo were diarrhea (7% Lipfendra versus 2% placebo) and dizziness (9% Lipfendra versus 4% placebo). Rates of treatment discontinuation due to adverse events were comparable between the treatment and placebo groups.
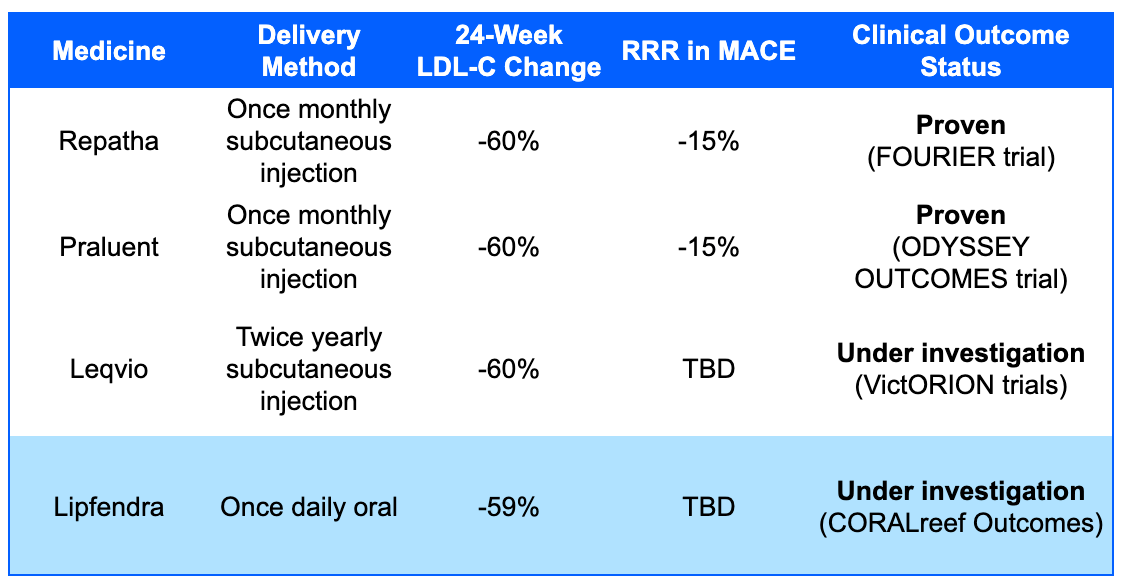
Sources: Merck press release, Lipfendra label
Celcuity - Revtorpyk (Pan-PI3K/mTOR inhibitor) / Approved (2L HR+ HER2- BC)
On July 14, 2026, the FDA approved Revtorpyk (gedatolisib), developed by Celcuity, as first and only FDA-approved therapy that inhibits all class I PI3K isoforms (α, β, δ, γ) for the treatment of adult patients with hormone receptor (HR)-positive, HER2-negative locally advanced or metastatic breast cancer without a PIK3CA mutation. Revtorpyk is indicated in combination with Faslodex (fulvestrant), with or without Ibrance (palbociclib), for patients whose disease has progressed following at least one line of endocrine therapy. This approval addresses a significant unmet need for the roughly 60% of HR+/HER2- breast cancer patients who are PIK3CA wild-type and have historically had fewer targeted treatment options after endocrine therapy resistance.
The approval was based on the Phase 3 VIKTORIA-1 trial, which showed that Revtorpyk significantly improved progression-free survival (PFS) compared to Faslodex alone in PIK3CA wild-type patients. We covered their ASCO 2026 presentation here in early June. Their triplet Regimen (Revtorpyk + palbociclib + fulvestrant) reduced the risk of disease progression or death by 76%. The most notable safety concern is stomatitis, which occurred in 72% of patients in the triplet group (22% Grade 3). Prophylactic use of a steroid-containing, alcohol-free mouthwash is required.
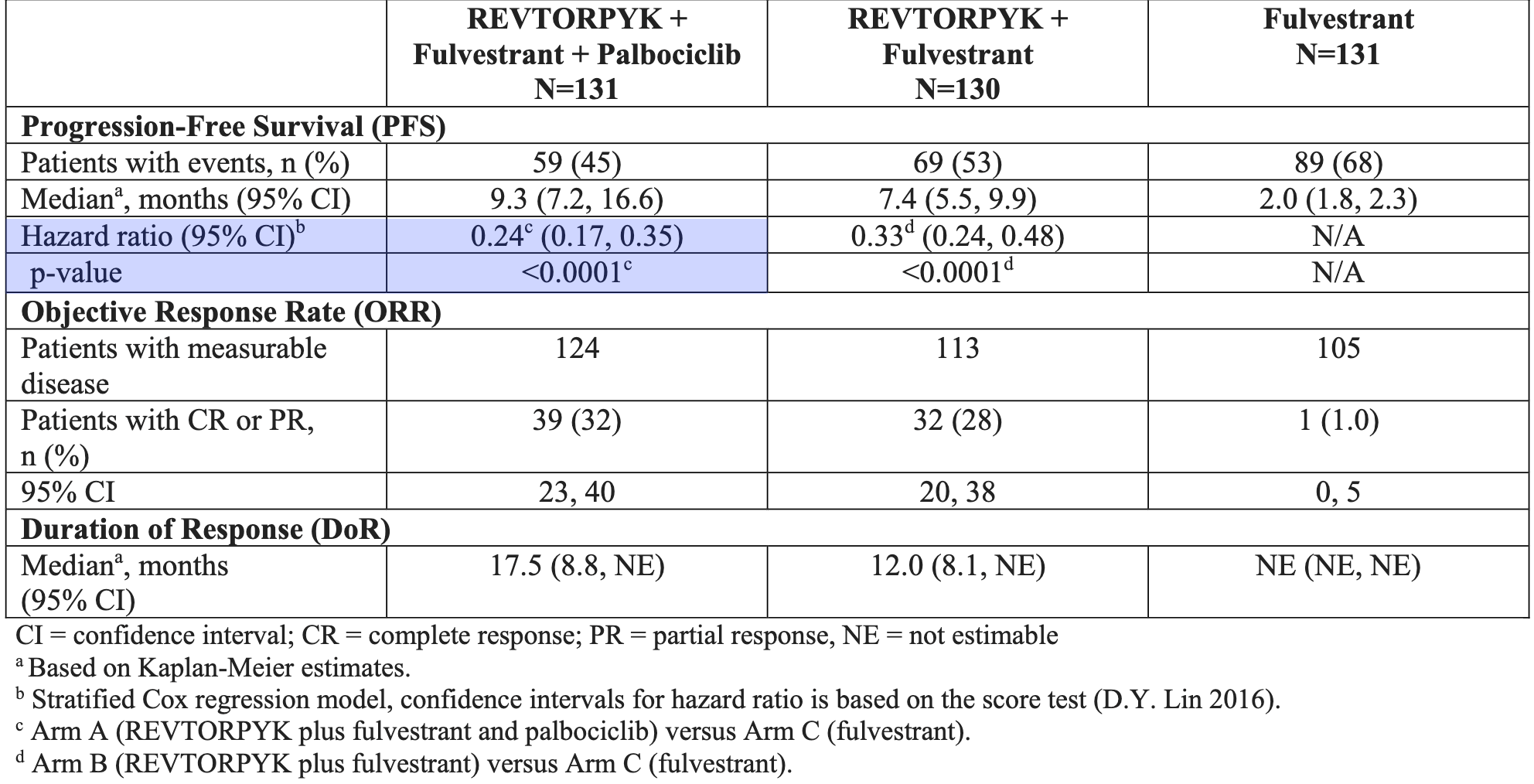
Separately, Celcuity has reported strong efficacy for Revtorpyk in patients with PIK3CA mutations, where it has demonstrated a 50% risk reduction in disease progression compared to Piqray (alpelisib) plus Faslodex (fulvestrant). Celcuity plans to submit a supplemental New Drug Application (sNDA) in 3Q 2026 to seek label expansion for this patient population. The company is also evaluating the drug in the Phase 3 VIKTORIA-2 trial as a potential first-line treatment.
Sources: Celcuity press release, Revtorpyk label
Label Expansions
Biogen - Leqembi IQKLIK (anti-Aβ mAb) / Approved (Alzheimer’s): On July 13, 2026, the FDA approved a label expansion for Leqembi IQLIK (lecanemab-irmb), an anti-amyloid monoclonal antibody for early Alzheimer’s disease, authorizing the use of a weekly subcutaneous autoinjector as an initiation dose for patients. Previously, this at-home, under-the-skin injection was only indicated as a maintenance therapy for patients who had already completed 18 months of intravenous infusions. This new approval allows patients and their caregivers to transition to home-based administration from the start of their treatment journey. Supported by clinical data demonstrating that subcutaneous administration achieves drug exposure and biomarker benefits equivalent to the IV formulation. This change could significantly reduce the logistical burden of recurring clinic visits and potentially provide greater flexibility for patients managing early-stage Alzheimer’s. Sources: Biogen press release, new Leqembi IQKLIK label
Pfizer - Padcev (Nectin-4 ADC) / Approved (MIBC): On July 10, 2026, the FDA approved a label expansion for the combination of Padcev (enfortumab vedotin-ejfv) and Keytruda (pembrolizumab) or the subcutaneous formulation Keytruda Qlex as a perioperative (neoadjuvant and adjuvant) treatment for adults with muscle-invasive bladder cancer (MIBC). Previously limited to cisplatin-ineligible patients, this expanded indication allows the use of the regimen for all patients with MIBC who are candidates for radical cystectomy, regardless of their cisplatin eligibility. The approval is supported by findings from the Phase 3 KEYNOTE-B15 (EV-304) trial, which demonstrated that this platinum-free combination significantly outperformed standard-of-care neoadjuvant chemotherapy, reducing the risk of disease progression, recurrence, or death by 47% and the risk of death by 35%, thereby establishing a new potential standard of care in the curative-intent setting. Sources: Pfizer press release, new Padcev label
Novartis - Fabhalta (factor B inhibitor) / Approved (IgAN): On July 17, 2026, the FDA granted traditional approval to Novartis’ Fabhalta (iptacopan) to slow the decline of kidney function in adults with primary immunoglobulin A nephropathy (IgAN) at risk of disease progression. This milestone converts the drug’s previous 2024 accelerated approval (was based on the surrogate endpoint of proteinuria reduction) into a full approval, supported by robust data from the Phase 3 APPLAUSE-IgAN clinical trial. Results from the trial demonstrated that patients treated with the oral Factor B inhibitor experienced a significantly slower annualized decline in estimated glomerular filtration rate (eGFR) compared to those on placebo, reinforcing the drug’s role in preserving long-term kidney health by targeting the alternative complement pathway. As part of its safety profile, Fabhalta remains available through a Risk Evaluation and Mitigation Strategy (REMS) program due to the risk of serious infections from encapsulated bacteria, requiring patients to be appropriately vaccinated prior to initiating treatment. Sources: Novartis press release
Clinical Trial Data
Q32 Bio - Bempikibart (anti-IL-7Rα mAb) / Phase 2a (alopecia)
On July 13, 2026, Q32 Bio announced positive topline results from Part B of the Phase 2a SIGNAL-AA clinical trial, evaluating bempikibart (ADX-914) in patients with severe or very severe alopecia areata (AA).
Severe alopecia areata is an autoimmune disease characterized by a loss of hair follicle immune privilege, leading to T-cell-mediated destruction and significant hair loss, currently managed by JAK inhibitors like Olumiant (baricitinib) and Litfulo (ritlecitinib) that suppress inflammatory signaling at the cost of potential systemic safety risks. Bempikibart offers a novel therapeutic approach as a fully human anti-IL-7Rα antibody, which differentiates itself from these broad-acting JAK inhibitors by specifically blocking IL-7 and TSLP signaling to re-regulate the adaptive immune system. This targeted mechanism aims to reset immune balance rather than simply suppressing inflammation, potentially providing a safer and more durable treatment alternative for patients who require systemic intervention for their condition.
In the Phase 2a SIGNAL-AA open-label trial, 33 patients with severe or very severe alopecia areata (baseline SALT score 50–100) received a 36-week regimen of bempikibart, demonstrating meaningful clinical efficacy with a mean 35.3% reduction in SALT scores and up to 30.3% of the ITT population achieving ≥20% scalp coverage (SALT-20). On a cross-trial basis, this 30% SALT-20 responder rate compares favorably to FDA-approved JAK inhibitors Olumiant (baricitinib) and Litfulo (ritlecitinib), which generally show SALT-20 response rates in the range of 30-40% in severe patient populations. The treatment was generally well-tolerated, with no treatment-related serious or Grade ≥3 adverse events, and only mild injection-site reactions reported. These results position bempikibart as a potentially disruptive, safer alternative to the currently dominant JAK inhibitor class, offering the possibility of a remittive effect without the systemic safety risks associated with chronic JAK-STAT pathway suppression.
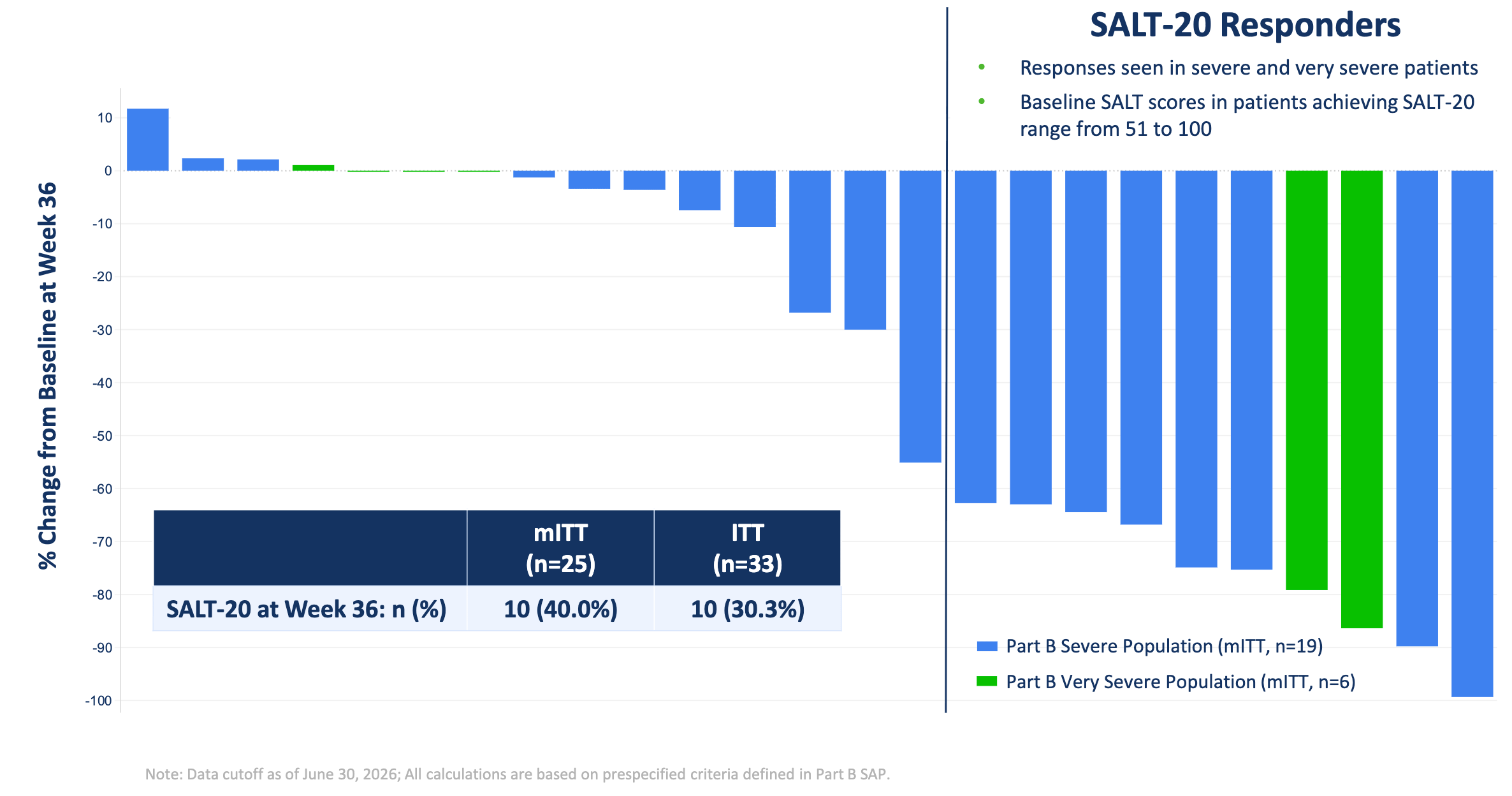
Q32 Bio intends to advance bempikibart into a pivotal clinical program in the first half of 2027. Full results from Part B will be presented at a future medical meeting. The company is also continuing to monitor patients through a 52-week off-drug follow-up and an ongoing open-label extension (OLE) study to further evaluate the durability of the treatment effect.
Sources: Q32 Bio press release, Q32 Bio slide deck
Veradermics - VDPHL01 (ER oral minoxidil) / Phase 2 (female androgenetic alopecia)
On July 15, 2026, Veradermics announced positive topline results from the Phase 2 Study ‘207’ (NCT06527365) evaluating VDPHL01, a proprietary extended-release oral minoxidil formulation, for the treatment of female pattern hair loss (androgenetic alopecia).
Female androgenetic alopecia is a progressive condition driven by genetic sensitivity to androgens that causes follicular miniaturization and thinning, typically managed with burdensome topical minoxidil or off-label hormonal therapies that often yield inconsistent results. VDPHL01 addresses this clinical gap as a novel, extended-release oral minoxidil formulation designed to stimulate hair regrowth by prolonging the anagen phase through sustained follicular exposure. By utilizing this specialized delivery technology, VDPHL01 aims to provide the efficacy of systemic minoxidil while mitigating the high plasma concentration spikes that cause cardiac side effects in immediate-release formulations, potentially offering a more convenient and safer oral alternative for long-term treatment.
In the Phase 2 Study ‘207’, 28 women with mild-to-moderate androgenetic alopecia treated with VDPHL01 demonstrated robust hair regrowth, achieving mean increases in non-vellus hair counts of 22.7 to 23.3 hairs/cm² and reporting high patient satisfaction, with approximately 90% noting improvements in hair coverage as early as two months into the trial. The formulation was well-tolerated with no treatment-related serious adverse events or cardiac issues of special interest, confirming that the extended-release technology successfully maintains efficacy while avoiding the systemic safety risks typical of immediate-release oral minoxidil. These proof-of-concept results establish VDPHL01 as a potentially best-in-class, non-hormonal oral therapy that addresses a significant unmet need for a more convenient and standardized treatment for female pattern hair loss.
Veradermics is currently enrolling patients in a registration-directed Phase 2/3 trial (Study ‘306’). The company anticipates topline results from the Phase 2/3 study in the first half of 2027, aims to further validate the drug’s potential for U.S. regulatory approval.
Sources: Veradermics press release
Biogen - Diranersen (tau ASO) / Phase 2 (Alzheimer’s)
On July 17, 2026, Biogen announced results from the Phase 2 CELIA trial of diranersen, an investigational antisense oligonucleotide (ASO) targeting tau protein, for the treatment of early-stage Alzheimer’s disease. Results were presented the Alzheimer’s Association International Conference (AAIC) 2026 in London, UK.
Alzheimer’s disease is a progressive neurodegenerative disorder driven by the accumulation of amyloid-beta plaques and tau-containing neurofibrillary tangles, which currently relies on symptomatic management alongside recently approved anti-amyloid monoclonal antibodies that provide modest clinical benefit by targeting amyloid clearance. Diranersen offers a distinct therapeutic approach as an antisense oligonucleotide (ASO) designed to intervene in this pathology by binding to the pre-mRNA of the MAPT gene to reduce tau protein expression. By specifically decreasing the production of this key component of neurofibrillary tangles, diranersen aims to address the tau-driven component of the disease, potentially offering a complementary or alternative strategy to existing treatments that primarily target amyloid pathology.
In the Phase 2 CELIA trial, patients with early-stage Alzheimer’s disease treated with intrathecal diranersen demonstrated a 26% reduction in clinical decline on the CDR-SB scale, a result numerically comparable to established anti-amyloid therapies like Leqembi and Kisunla. Despite this efficacy signal, the trial revealed an unexpected inverse dose-response relationship, where the lowest dose provided the most robust benefit while higher doses showed diminishing returns. This finding could pose a significant regulatory challenge, necessitating further investigation into whether higher levels of tau knockdown trigger compensatory mechanisms or off-target effects, and highlighting the critical need to identify the optimal biological window for therapeutic tau modulation in future development.
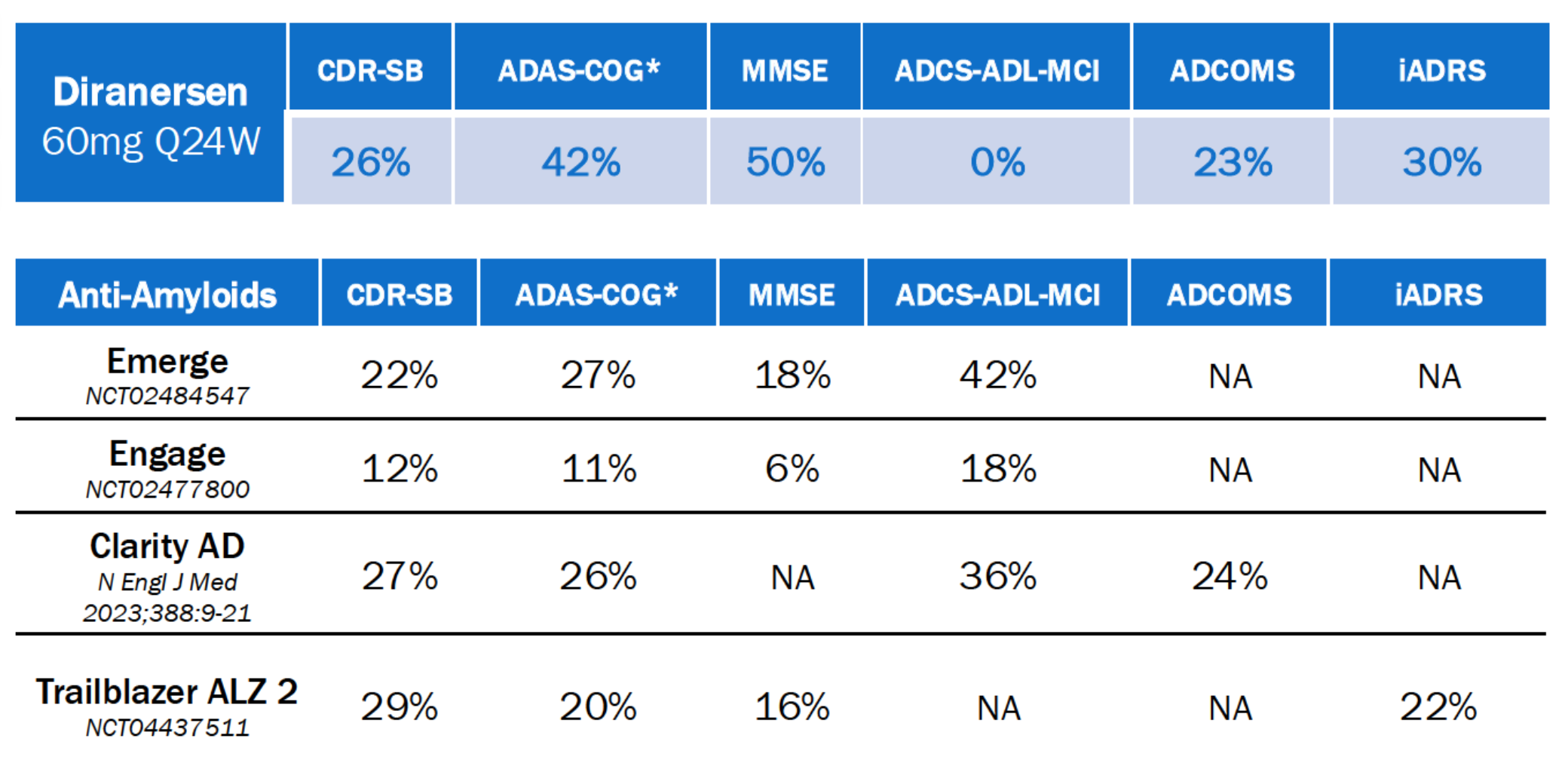
Biogen plans to conduct extensive post-hoc analyses. The company has not yet provided a definitive timeline for a Phase 3 transition. Future studies may need to focus on refining the dosing regimen and identifying the specific patient sub-populations most likely to benefit from this precise level of tau reduction.
Sources: Biogen press release, Biogen AAIC 2026 slide deck
Ipsen - Iqirvo (PPARa/d dual agonist) / Phase 3b (PBC)
On July 13, 2026, Ipsen announced positive topline results from the Phase 3b ELSPIRE trial, which evaluated the efficacy and safety of Iqirvo (elafibranor) in patients with primary biliary cholangitis (PBC).
Primary biliary cholangitis (PBC) is a chronic, autoimmune-mediated cholestatic disease that destroys small intrahepatic bile ducts, causing bile acid accumulation, inflammation, and potential progression to cirrhosis, making the normalization of biomarkers like alkaline phosphatase (ALP) the primary target to improve patient outcomes. While ursodeoxycholic acid (UDCA) serves as the first-line standard of care, second-line treatments are essential for patients who remain at risk due to inadequate responses. Iqirvo (elafibranor) addresses this need as an oral, once-daily dual PPAR α and δ agonist that modulates bile acid synthesis, detoxification, and transport to decrease bile toxicity and provide anti-inflammatory and anti-fibrotic effects, thereby offering a more targeted approach to achieving biochemical normalization and slowing liver injury.
The Phase 3b ELSPIRE trial demonstrated the therapeutic potential of Iqirvo in early-stage primary biliary cholangitis by enrolling 92 patients with modest baseline alkaline phosphatase (ALP) elevations (1-1.67x ULN) who had an inadequate response to ursodeoxycholic acid. Patients randomized to once-daily Iqirvo showed statistically significant superiority, with 85% achieving ALP normalization (≤1 ULN) at 52 weeks compared to only 23% in the placebo group, all while maintaining a consistent and favorable safety profile. These findings are highly significant as they confirm that achieving optimal biochemical normalization is feasible in a less severe disease population, providing the clinical evidence needed to support a potential label expansion that could facilitate earlier therapeutic intervention and potentially improve long-term patient prognosis.
Ipsen plans to submit these data to regulatory authorities (including the FDA and EMA) to support the expanded use of Iqirvo. The company intends to present the full results from the ELSPIRE trial at an upcoming medical meeting.
Sources: Ipsen press release
Descriptive data releases without numerical data
Takeda - Zasocitinib (TYK2 inhibitor) / Phase 3 (psoriasis): In Phase 3 trials (LATITUDE PsO 3001 and 3002), Takeda’s once-daily oral TYK2 inhibitor, zasocitinib, demonstrated robust and consistent skin clearance across the body, including historically difficult-to-treat areas. Building upon topline results announced in December 2025 and data presented in March 2026, these secondary endpoint results revealed that approximately 75% of patients with scalp psoriasis and 70% of those with palmoplantar disease achieved clear or almost clear skin (ssPGA/hfPGA 0/1) at week 16, significantly outperforming both placebo and the active comparator apremilast. Furthermore, the drug showed statistically significant improvements in nail psoriasis as measured by the NAPSI score (scalp, nails, hands and feet), with benefits across these high-impact sites sustained through week 24, reinforcing zasocitinib’s potential to offer meaningful, whole-body skin clearance as a leading oral treatment option for moderate-to-severe plaque psoriasis. Sources: Takeda press release
Merck - Sac-TMT (TROP2 ADC) / Phase 3 (1L PDL1-negative metastatic non-sq NSCLC): In the Phase 3 OptiTROP-Lung06 trial, the combination of sacituzumab tirumotecan (sac-TMT), a TROP2-directed antibody-drug conjugate (ADC), and Keytruda (pembrolizumab) achieved its primary endpoint by demonstrating a statistically significant and clinically meaningful improvement in progression-free survival (PFS) compared to the standard-of-care regimen of pembrolizumab plus platinum-based chemotherapy in patients with previously untreated, PD-L1-negative, metastatic non-squamous non-small cell lung cancer (NSCLC). These results, announced by Kelun-Biotech on July 14, 2026, represent the first instance of an ADC-plus-immunotherapy regimen successfully replacing conventional platinum-doublet chemotherapy in this difficult-to-treat population, where checkpoint inhibitors alone typically demonstrate limited benefit. In addition to the significant PFS improvement, a positive trend in overall survival was observed, and the safety profile remained consistent with prior studies. Sources: Kelun press release
GSK - Jemperli (anti-PD1 mAb) / Phase 3 (Stage 2/3 dMMR/MSI-H rectal cancer): In the registrational Phase 2 AZUR-1 trial, GSK’s anti-PD-1 monoclonal antibody Jemperli (dostarlimab) demonstrated a clinically meaningful and sustained clinical complete response (cCR) rate at 12 months in patients with stage II/III mismatch repair-deficient/microsatellite instability-high (dMMR/MSI-H) locally advanced rectal cancer. By meeting this primary objective, the data support the potential for Jemperli to become the first immunotherapy capable of enabling some patients to avoid the significant morbidity associated with traditional trimodality therapy, specifically the long-term impacts of chemotherapy, radiation, and surgery. The interim analysis confirmed that the safety and tolerability profile of the drug remains consistent with its established performance in other solid tumors, with no new safety signals identified. GSK plans to share these results with global regulatory authorities, including for accelerated review in the United States, to potentially transform the standard of care for this biomarker-defined population. Sources: GSK press release
Love biotech? Check out Biotech Readout’s full content library, or navigate directly to a segment that interests you:
Frontiers in Medicine: Exploring the frontiers of our understanding and treatments for disease.
Medical History: Recovering forgotten relics in the history of medicine.
Acquisitions: Exploring the innovation behind acquired companies.
Weekly Readout: A digest of new clinical data from the past week.

To contact us, please send us an email at biotechreadout@gmail.com
Disclaimers
Investigational Status Disclaimer
The therapeutic candidates discussed in this newsletter are currently in clinical development and have not been approved for commercial sale by the U.S. Food and Drug Administration (FDA), the European Medicines Agency (EMA), or other global regulatory authorities. Their safety and efficacy have not been established. References to pipeline products and ongoing clinical trials involve significant risks and uncertainties. Statements regarding the potential safety, potency, or efficacy of investigational drugs reflect current hypotheses and are not a guarantee of future performance or regulatory clearance. The outcome of clinical trials is inherently unpredictable, and clinical results from earlier stages may not be predictive of results in later, larger-scale trials.
No Medical Advice Disclaimer
This newsletter is for informational and educational purposes only. The content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or another qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read in this publication.
No Patient-Provider Relationship Disclaimer
The information provided in this newsletter is for educational and analytical purposes only. Receipt of this information, or any interaction with this content, does not create a physician-patient, pharmacist-patient, or any other professional-provider relationship between you and the authors or publishers. This newsletter should not be used as a substitute for a personal consultation with a qualified healthcare professional.
Forward-Looking Statements Disclaimer
This newsletter contains “forward-looking statements” regarding future events, including clinical trial timing, regulatory milestones, and projected market performance. These statements are based on current expectations and assumptions that are subject to significant risks and uncertainties. Actual results may differ materially from those expressed or implied. We undertake no obligation to update these statements as a result of new information or future developments.
Third-Party Links & Content Disclaimer
This newsletter contains links to third-party websites, including clinical trial registries and corporate presentations. Biotech Readout does not endorse, guarantee, or assume responsibility for the accuracy or reliability of any information offered by third-party providers.
Errors and Omissions Disclaimer
While we strive for technical accuracy, the information in this newsletter is provided on an “as is” basis with no guarantees of completeness, accuracy, or timeliness. Biotech Readout assumes no liability for any errors or omissions in the content of this publication.
Non-Endorsement Disclaimer
Any reference to specific commercial products, processes, or services by trade name, trademark, or manufacturer does not constitute or imply an endorsement or recommendation by the author. All trademarks are the property of their respective owners.
No Investment Advice Disclaimer
This newsletter is for informational purposes only and does not constitute financial, investment, or legal advice. The author is not a registered investment advisor. You should consult with a professional financial advisor before making any investment decisions. The biotechnology sector is highly volatile; past performance is not indicative of future results.
Conflict of Interest Disclaimer
The author of this newsletter maintains a position of independence. At the time of publication, the author holds no direct financial interest, equity, or options in any of the companies mentioned in this report. No compensation has been received from any third party to feature or analyze specific therapeutic candidates or corporate entities.