
Obesity drugs for cancer, deleting cholesterol, Hepatitis B functional cure, and more
Weekly Readout #9: Week ending May 29, 2026

Disclaimer: This newsletter is for educational and informational purposes only and does not constitute medical, investment, or financial advice, nor does it establish a provider-patient relationship. Content may include forward-looking statements and discussions of investigational therapeutic candidates that are not FDA/EMA approved; their safety and efficacy remain unestablished and clinical outcomes are unpredictable. While we strive for accuracy, all information is provided as is without guarantees. This newsletter is independent, and the author holds no financial positions in the companies mentioned nor receives third-party compensation for this coverage. Please find a complete version of our disclaimers at the bottom of this article and on our About page.

Press ⬇️ to go down to a section and ⬆️ to go back up to the Table of Contents
Table of Contents
This week, we discuss…
…novel scientific findings with immediate implications in drug development:
Potentially reducing the aggressiveness of 6 types of cancer with GLP1 obesity medicines; GLP-1 receptor agonists / Real-world data study (7 cancer types) ⬇️
Dueling datasets in gene therapies for potentially long-lasting cholesterol reduction; Eli Lilly/Verve - VERVE-102 (PCSK9 KO) / Phase 1b (HeFH) & Editas - EDIT-401 (LDLR upregulator) / Preclinical data (ASCVD) ⬇️
…potentially first-in-class or best-in-disease treatments:
Hepatitis B functional cure; GSK - Bepirovirsen (HBV RNA ASO) / Phase 3 (Hepatitis B) ⬇️
Numerically higher progression-free survival compared to PD1 x VEGF bsAb; Merck & Kelun - sac-TMT (TROP2 ADC) / Phase 3 (NSCLC) ⬇️
Apogee - Zumilokibart (anti-IL13 mAb) / Phase 2 (atopic dermatitis) ⬇️
Arrowhead - ARO-INHBE (INHBE RNAI) / Phase 1/2a (obesity & MASH) ⬇️
GLP-1 receptor agonists / Real-world data study (6 cancer types)
An ASCO abstract shifts the focus to a fascinating trend in modern oncology: exploring whether glucagon-like peptide-1 receptor agonists (GLP-1RAs), like semaglutide or tirzepatide, exert protective, antineoplastic effects when initiated after a cancer diagnosis. This research was conducted by a team of investigators from the Cleveland Clinic. The lead study author who presented the findings is Dr. Mark David Orland, MD, a physician at the Cleveland Clinic. His co-authors on the study include A. Mandala, S. Unlu, and colleagues. Shout out to The M&A Hunter, who brought this abstract to my attention and has a great writeup on it, linked here.
Indication: 6 cancer types.
Mechanism: While GLP-1 receptor agonists are traditionally utilized for glycemic control in type 2 diabetes and weight management in obesity, emerging preclinical and observational oncology data suggest they possess pleiotropic, immunomodulatory, and direct anti-neoplastic properties. GLP-1RAs are hypothesized to reduce chronic, systemic, and tissue-specific inflammation, a well-known driver of tumor progression and metastasis. GLP-1 receptors (GLP-1R) are expressed natively on certain epithelial tissues and solid tumors. Ligand binding can potentially downregulate downstream oncogenic signaling pathways (such as PI3K/AKT/mTOR or MAPK/ERK) that govern cell proliferation. By improving insulin sensitivity and reducing circulating levels of insulin and insulin-like growth factors (IGFs), which function as potent mitogens, GLP-1RAs may indirectly reduce the availability of metabolic growth signals to tumor cells.
Trial design: This was a large-scale, retrospective, propensity score-matched cohort study leveraging real-world data to evaluate the clinical impact of GLP-1RA exposure. Patients diagnosed with one of seven major solid malignancies (including non-small cell lung cancer [NSCLC], breast, colorectal [CRC], and hepatocellular carcinoma [HCC]). The study compared patients who initiated a GLP-1RA following their definitive cancer diagnosis versus propensity-matched control patients with similar baseline demographics, metabolic comorbidities (e.g., type 2 diabetes, BMI), and cancer stages who did not receive GLP-1RAs. The primary outcome was the risk of progression to metastatic disease. Additionally, investigators evaluated the correlation between a tumor’s baseline GLP-1R expression levels and overall survival (OS).
Data:
Efficacy: The study revealed a striking correlation between post-diagnostic GLP-1RA use and reduced tumor aggressiveness. Post-diagnostic GLP-1RA exposure was associated with a reduced rate of metastatic progression across 6 out of the 7 solid malignancies evaluated. The risk reductions reached high statistical significance in four specific primary cancer types: NSCLC, Breast Cancer, Colorectal Cancer (CRC), and Hepatocellular Carcinoma (HCC). High baseline expression of the GLP-1 receptor within tumor tissue independently predicted superior survival outcomes across all seven tumor types combined (HR = 0.67, 95% CI: 0.54–0.83, p < 0.001). This effect was most pronounced in breast cancer, where high GLP-1R expression yielded a 45% relative reduction in the risk of death (HR = 0.55, 95% CI: 0.35–0.87, p = 0.011).
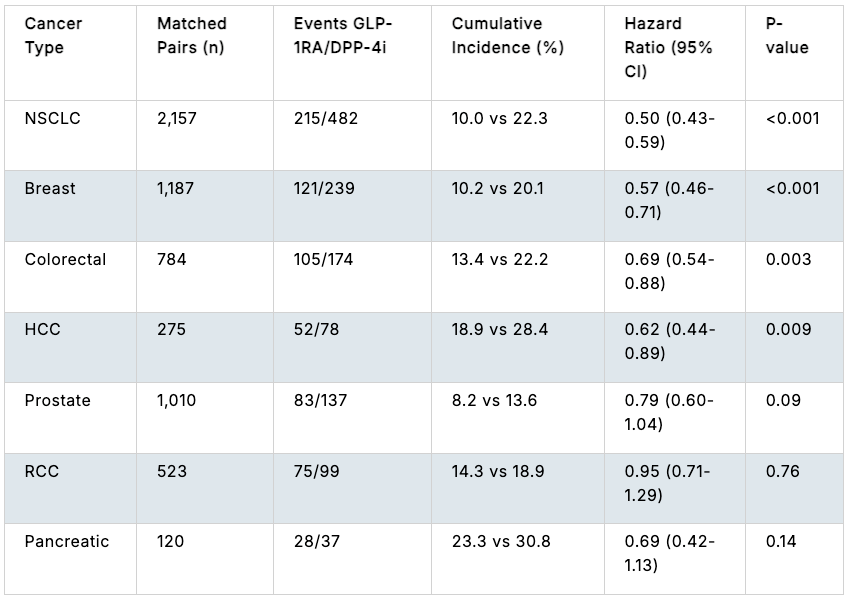
Effect of GLP1-1RA exposure compared to DPP-4i exposure on progression of stage I-III cancer to stage IV cancer; Source: ASCO abstract Safety: No adverse safety signals or increases in unexpected toxicities were seen in the GLP-1RA cohort compared to the matched controls.
Impact:
Beyond Glycemic Control: It suggests that the benefits of GLP-1RAs in cancer patients extend far beyond managing diabetes or weight; they may actively alter the biology of the tumor’s microenvironment to suppress secondary seeding (metastasis).
Next steps: Since these findings are derived from a retrospective, propensity-matched database, they are subject to residual confounding. The investigators emphasize that these results warrant immediate validation in prospective, randomized controlled trials (RCTs). No such clinical trials are currently being conducted by Eli Lilly (tirzepatide) or Novo Nordisk (semaglutide). Basic science and translational studies are urgently needed to fully map out the precise molecular, intracellular, and immunological pathways driven by GLP-1R activation in malignant tissues. Future research will likely explore whether combining GLP-1RAs with standard cytoreductive therapies (like chemotherapy, targeted agents, or immune checkpoint inhibitors) can synergistically delay or prevent distant recurrence in high-risk, early-stage patients.
Dueling Datasets
A comparison between Eli Lilly/Verve’s VERVE-102 and Editas Medicine’s EDIT-401 highlights the front lines of genetic cardiovascular medicine. It highlights a comparative look at two distinct modalities, Adenine Base Editing (ABE) vs. CRISPR-Cas9 Nuclease, and two fundamentally different biological strategies: gene knockout vs. gene upregulation. While VERVE-102 has successfully demonstrated human proof-of-concept, EDIT-401 has shown massive lipid-lowering potential in non-human primates (NHPs).
Eli Lilly/Verve - VERVE-102 (PCSK9 KO) / Phase 1b (HeFH)
The positive Phase 1b interim data readout for VERVE-102, an investigational in vivo adenine base-editing therapy developed by Verve Therapeutics and acquired by Eli Lilly, was presented at the European Atherosclerosis Society (EAS) Congress and simultaneously published in The New England Journal of Medicine (NEJM).
Editas - EDIT-401 (LDLR upregulator) / Preclinical data (ASCVD)
The highly anticipated preclinical data for EDIT-401, an investigational in vivo CRISPR gene-editing medicine developed by Editas Medicine, was presented in oral sessions at major medical congresses the 94th European Atherosclerosis Society (EAS) Congress.
Indication: Atherosclerotic Cardiovascular Disease (ASCVD) develops as circulating apolipoprotein B (ApoB)-containing lipoproteins, primarily low-density lipoprotein cholesterol (LDL-C), penetrate the arterial intima, undergo oxidation, and trigger a chronic, macrophage-driven inflammatory response that forms fibrofatty plaques prone to thrombosis, myocardial infarction, and stroke. Heterozygous Familial Hypercholesterolemia (HeFH) dramatically accelerates this process. It is an autosomal dominant genetic disorder caused by mutations in the LDLR, APOB, or PCSK9 genes that impair hepatic LDL receptor function, leading to lifelong, severely elevated baseline LDL-C levels (≥190 mg/dL) and an exponentially higher lifetime cumulative burden of atherogenic exposure. According to the 2026 ACC/AHA multisociety guidelines, the standard of care for secondary prevention in ASCVD and for high-risk HeFH mandates aggressive lipid-lowering therapy to hit strict target thresholds (e.g., LDL-C<55 mg/dL for very high-risk ASCVD and <70 mg/dL for primary prevention HeFH). This is achieved through a foundation of intensive lifestyle optimization paired with a sequential pharmacological escalation strategy, beginning with maximum-dose high-intensity statins, followed by the timely addition of ezetimibe, and progressing to injectable biologics like PCSK9 monoclonal antibodies (e.g., evolocumab, alirocumab) or siRNA therapies (inclisiran) when targets are not met.
Mechanism:
VERVE-102 (PCSK9 Knockout Approach): This therapy uses an in vivo Adenine Base Editing (ABE), an advanced form of CRISPR that alters a single base pair without double-stranded DNA breaks. It introduces a single-nucleotide change (A-T to G-C) to create a premature stop codon, turning off the PCSK9 gene in liver cells. This prevents the degradation of LDL receptors (LDLR), keeping them on the cell surface to clear cholesterol.
EDIT-401 (LDLR Upregulation Approach): This therapy uses an in vivo CRISPR-Cas9 nuclease, a gene-editing technique that creates precise double-stranded cuts. It cuts and permanently deletes the genetic “brakes” that destabilize LDLR mRNA, specifically the negative regulatory element in the 3’ UTR region. By removing this region, it forces a ≥6-fold increase in functional LDL receptor expression directly, bypassing the need to target upstream proteins.
Trial design: Since these programs are at different stages of clinical development, we are comparing VERVE-102’s Phase 1b human data against EDIT-401’s preclinical NHP data.

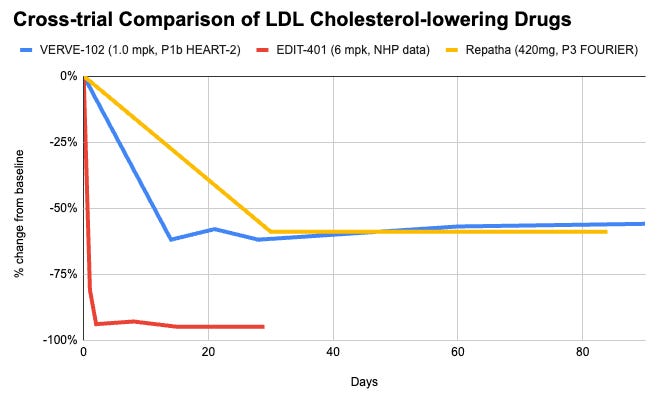
Data (Eli Lilly press release, NEJM paper, Editas press release, Editas slide deck):
Efficacy: VERVE-102 is biologically capped by how much cholesterol can be cleared simply by removing PCSK9. A ~60% reduction in LDL-C matches the maximum therapeutic effect seen with commercial PCSK9 monoclonal antibodies (e.g., Repatha, Praluent) or siRNA (Leqvio). It just delivers it via a permanent, single-dose solution. EDIT-401 targets the final common pathway of lipid clearance: the LDL receptor itself. By removing the genetic brakes to overexpress LDLR, it demonstrated an unprecedented 95% reduction in LDL-C in non-human primates. Furthermore, because it increases raw receptor density, it simultaneously results in an up to 92% reduction in Lipoprotein(a), a highly atherogenic and historically hard-to-drug particle that standard PCSK9 strategies struggle to clear to this magnitude.
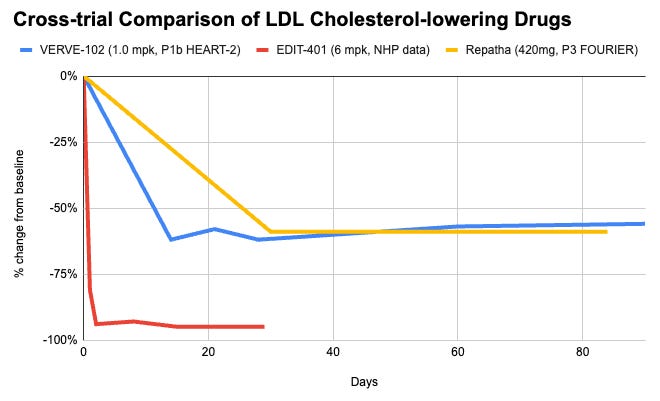
Safety: VERVE-102 uses Base Editing, which does not cut the DNA backbone. This significantly minimizes the risk of insertions, deletions (indels), chromosomal translocations, or complex genomic rearrangements. This gives it a preferred safety profile for large-scale chronic diseases. EDIT-401 utilizes standard Cas9 Nuclease to make a double-stranded break using dual guide RNAs. While effective at knocking out the 3’ UTR regulatory region in models to date, double-stranded breaks introduce potential risks of off-target chromosomal toxicity or structural variants, which require long-term clinical safety evaluation in human subjects.
Impact:
Potential for on-and-done, cholesterol lowering gene therapies: In real-world clinical practice, long-term adherence to daily oral therapies or bi-weekly injections is notoriously poor due to pill fatigue, side effects, costs, and systemic healthcare inequities. Since cardiovascular disease is a silent, cumulative process, poor compliance leads directly to preventable major adverse cardiac events (MACE). A single-dose, long-lasting therapeutic approach is theorized to shift the treatment paradigm from chronic disease management toward a durable genetic modification, ensuring lifelong, flawless lipid control regardless of human behavior. Given the risk of liver toxicity, the jury is still out on whether these will ultimately capture meaningful market share in the therapeutic landscape, and requires large & comprehensive placebo-controlled trials and regulatory review. View on these therapies are split along two lines: those who are more excited about the efficacy and those who are more cautious about its potential toxicity.


Next steps: The next steps for both programs focus on critical 2026 clinical transitions. Eli Lilly and Verve are finalizing the dose-escalation portion of their Phase 1b Heart-2 trial for VERVE-102 and are on track to initiate a broader Phase 2 clinical study in patients with HeFH and premature coronary artery disease by the end of this year. Concurrently, Editas Medicine is transitioning EDIT-401 from preclinical validation to its first-in-human clinical trial; the company plans to submit a Clinical Trial Notification in Australia by mid-2026, launch a Phase 1 trial in severe HeFH patients in the second half of the year, and release early human proof-of-concept data by the end of 2026, with full topline results expected in 2027.
GSK - Bepirovirsen (HBV RNA ASO) / Phase 3 (Hepatitis B)
The positive, pivotal Phase 3 data readout for Bepirovirsen (GSK3228836), an investigational antisense oligonucleotide (ASO) co-developed by GSK and Ionis Pharmaceuticals, was presented in a late-breaking session at the European Association for the Study of the Liver (EASL) Annual Congress and simultaneously published in The New England Journal of Medicine (NEJM). This historic readout establishes bepirovirsen as the first finite-duration therapy to successfully demonstrate high, statistically significant functional cure rates in a global Phase 3 program.
Indication: Chronic Hepatitis B (CHB) is a viral infection of the liver caused by the Hepatitis B virus (HBV), a partially double-stranded DNA virus that integrates into the host genome and establishes a stable transcriptional template called covalently closed circular DNA (cccDNA) inside hepatocyte nuclei. The resulting liver injury, cirrhosis, and hepatocellular carcinoma (HCC) are not directly cytopathic but are driven by a chronic, host-mediated immune response where cytotoxic T lymphocytes continuously destroy infected hepatocytes, leading to necroinflammation and progressive fibrogenesis. Progression is clinically monitored via phases dictated by viral load (HBV DNA), serological markers like HBeAg, HBsAg, and liver enzyme levels (ALT). According to AASLD and EASL clinical practice guidelines, the standard of care centers on lifelong, daily oral nucleos(t)ide analogues (NAs), principally tenofovir alafenamide (TAF), tenofovir disoproxil fumarate (TDF), or entecavir (ETV), which possess a high barrier to resistance and effectively suppress viral replication to undetectable levels, thereby halting disease progression and reducing HCC risk. However, because NAs fail to eliminate the intrahepatic cccDNA reservoir, functional cure (defined as sustained serum HBsAg loss with or without anti-HBs seroconversion) is achieved in fewer than 1–2% of patients, driving an aggressive industry pipeline focused on novel multi-drug regimens, including siRNA, antisense oligonucleotides (ASOs), and capsid assembly modulators (CAMs), designed to achieve functional cure after a finite treatment course.
Mechanism: Bepirovirsen is a systemically administered, unconjugated antisense oligonucleotide designed to clear chronic hepatitis B virus (HBV) through a distinctive multimodal mechanism. It is engineered to sequence-specifically bind to all HBV messenger RNA (mRNA) transcripts and pre-genomic RNA (pgRNA). Upon binding, it recruits cellular RNase H to cleave and degrade the viral transcripts. By destroying viral RNA templates, it systematically shuts down the synthesis of all downstream viral elements, most notably HBsAg (hepatitis B surface antigen). HBsAg is the primary decoy molecule the virus uses to exhaust and blind host immune responses. Collapsing circulating HBsAg concentrations eliminates the chronic antigen-overload burden, effectively lifting the immune-checkpoint “brakes” and allowing endogenous cytotoxic T lymphocytes and B cells to reactivate and eliminate remaining infected hepatocytes.
Trial design: The data represents the integrated and pooled results from two identical global, multi-center, randomized, double-blind, placebo-controlled Phase 3 confirmatory studies: B-Well 1 (NCT05630807) and B-Well 2 (NCT05630820). They evaluated 1,834 non-cirrhotic adults with chronic hepatitis B (CHB) who were already stable and virologically suppressed on background standard-of-care nucleos(t)ide analogue (NA) therapy (e.g., tenofovir, entecavir). Enrolled patients possessed low-to-moderate antigen loads, defined by baseline HBsAg ≤ 3,000 IU/mL. Patients were randomized 2:1 to add either a finite 24-week course of subcutaneous bepirovirsen (300 mg) or matching placebo to their stable background NA regimen. If patients met strict unmeasurable HBsAg and HBV DNA criteria between weeks 24 and 46, oral NA therapy was stopped at week 48. The primary endpoint was the proportion of patients achieving a functional cure (FC) at Week 72 (6 months after discontinuing all treatments), defined as qualitative HBsAg not detected (below 0.05 IU/mL) and serum HBV DNA below the lower limit of quantification (< 20 IU/mL) sustained for at least 24 consecutive weeks off all therapy.
Data (press release, slide deck, NEJM paper):
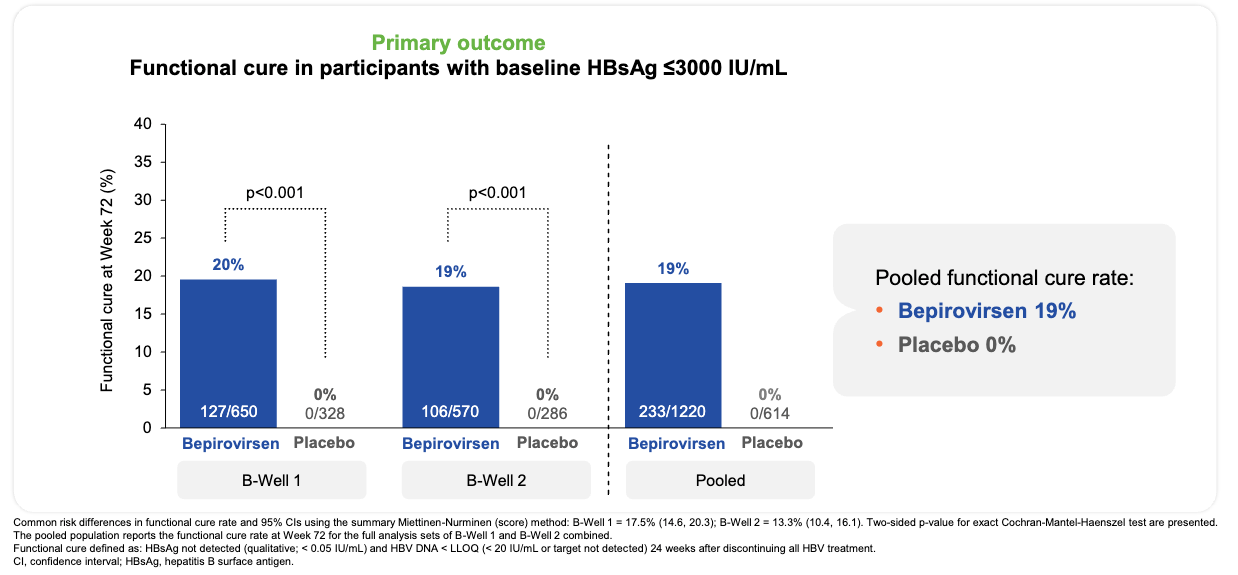
Efficacy: The trials met their primary and ranked secondary endpoints with high statistical significance, significantly shifting the landscape of finite hepatitis B treatment. In the overall population (HBsAg ≤ 3,000 IU/mL), 24 weeks of bepirovirsen achieved a 19% pooled functional cure rate (233 of 1,220 patients) across both trials (20% in B-Well 1, 19% in B-Well 2) versus 0% in the placebo arms (p < 0.001). Additionally, 49% of all bepirovirsen recipients achieved ultra-low residual surface antigen levels one full year after completing their treatment course (qHBsAg ≤ 100 IU/mL).
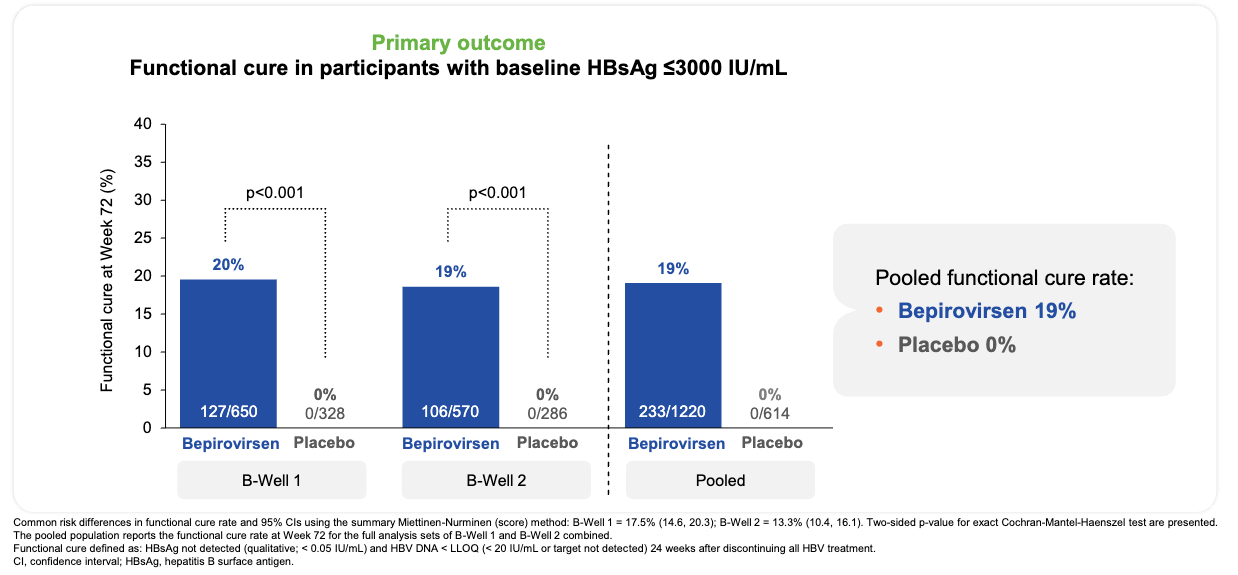
Functional cure was achieved in 19% of bepirovirsen recipients and no placebo recipients; Source: GSK presentation, slide 15 Safety: The most frequent adverse events were injection site erythema (31%) and local pain (23%). Transient elevations in alanine aminotransferase (>10x ULN) occurred in 6% of bepirovirsen patients, typically clustering between weeks 5 and 10. Crucially, these flares coincided directly with maximum antigen drops and represented successful immune-mediated clearance of infected hepatocytes; no cases were adjudicated as drug-induced liver injury (DILI). Discontinuations due to adverse events occurred in only 3% of the active treatment arm.
Impact:
Potentially best-in-class: Current standard-of-care oral NAs are highly effective at suppressing viral replication but achieve a functional cure in less than 1% of patients annually, locking patients into a lifetime of daily pills. Bepirovirsen’s 19–26% cure rate establishes a valid pathway toward a finite, 6-month treatment regimen.
Substantial Reduction in Long-Term Mortality: In hepatology literature, successful HBsAg loss and functional cure correlate with an 89% relative reduction in the risk of developing hepatocellular carcinoma (HCC) and a 62% reduction in all-cause mortality, dramatically altering the health trajectory for millions of individuals living with CHB.
Next steps: Regulatory filings are under advanced review across major global territories. The US FDA accepted the New Drug Application (NDA) for Priority Review with Breakthrough Therapy and Fast Track designations, setting a PDUFA action date of October 26, 2026. Parallel reviews are tracking via expedited pathways in Europe, Japan (SENKU designation), and China. GSK is targeting potential initial regulatory decisions as early as Q3 2026. GSK has kicked off its primary launch preparation steps, notably sealing a major strategic commercial collaboration with Sino Biopharmaceutical to rapidly scale patient diagnostics and access in China, which accounts for a massive proportion of the global chronic HBV burden. While the B-Well trials successfully cleared the path for low-to-moderate antigen cohorts, GSK is running the ongoing Phase IIb B-United clinical trial. This study evaluates combination regimens (including pairing bepirovirsen with immunomodulators like interferon) to try to drive equivalent functional cure rates in complex patients entering therapy with high baseline surface antigen levels (>3,000 IU/mL).
Merck & Kelun - sac-TMT (TROP2 ADC) / Phase 3 (NSCLC)
Indication: Non-small cell lung cancer (NSCLC) primarily arises from epithelial cell mutations within the central airways (squamous cell carcinoma) or peripheral lung tissue (adenocarcinoma), driven by chronic oncogenic insults like tobacco smoke or specific driver mutations (e.g., EGFR, ALK, ROS1). These genetic aberrations trigger dysregulated intracellular signaling cascades, bypass apoptosis, and promote a highly immunosuppressive tumor microenvironment. For advanced or metastatic disease without actionable driver mutations, the standard of care for first-line treatment relies on immune checkpoint inhibitors targeting the PD-1/PD-L1 pathway, either administered as a monotherapy for tumors with high PD-L1 expression (Tumor Proportion Score ≥50%) or combined with a platinum-doublet chemotherapy regimen (such as pembrolizumab plus carboplatin and pemetrexed for non-squamous histology) to elicit rapid cytoreduction alongside sustained systemic immune activation.
Mechanism: Sacituzumab Tirumotecan (sac-TMT / MK-2870 / SKB264) is an innovative, next-generation TROP2-directed antibody-drug conjugate (ADC) featuring a highly engineered molecular design. It utilizes a humanized IgG1 monoclonal antibody that selectively binds to TROP2 (trophoblast cell-surface antigen 2), a glycoprotein highly overexpressed across various epithelial tumors, including squamous and non-squamous NSCLC. It employs a unique, stable yet tumor-cleavable linker designed to balance systemic stability (preventing premature payload release in circulation) with efficient intratumoral release. The payload is a potent, belotecan-derived topoisomerase I inhibitor (similar to SN-38). Upon internalization, it causes DNA damage and cell death. The membrane-permeable nature of the freed payload allows it to diffuse out of the primary target cell and eradicate neighboring, heterogeneous tumor cells regardless of their TROP2 expression levels. Combining the ADC with an anti-PD-1 therapy (Keytruda) is hypothesized to leverage immunogenic cell death (ICD) triggered by the topoisomerase I payload, potentially upregulating tumor antigen presentation to support a T-cell-mediated immune response.
Trial design: The Chinese Phase 3 OptiTROP-Lung05 study involves the combination of sac-TMT plus pembrolizumab (Keytruda) compared to pembrolizumab monotherapy in patients with first line (1L) NSCLC. The primary endpoint is Progression-Free Survival (PFS) as assessed by a Blinded Independent Central Review (BICR). Key secondary endpoints include Overall Survival (OS), Objective Response Rate (ORR), Duration of Response (DoR), and Safety/Tolerability.
Data (ASCO abstract):
Efficacy: The trial met its primary endpoint with highly statistically significant and clinically meaningful efficacy improvements. The combination of sac-TMT + Keytruda demonstrated a 65% Relative Risk Reduction (RRR) for disease progression or death compared to Keytruda alone (expressed as a Hazard Ratio, HR = 0.35). The combination arm yielded a substantially elevated Objective Response Rate (ORR) and deeper, more durable responses relative to the anti-PD-1 monotherapy cohort.
Safety: The overall safety was “manageable and consistent with prior studies of both components”, according to the trial sponsor. The most common Grade ≥3 treatment-related adverse events (TRAEs) were hematologic, specifically anemia, leukopenia, and neutropenia. Crucially, sac-TMT continues to show a differentiated safety profile from other TROP2 ADCs, demonstrating a significantly lower incidence of severe interstitial lung disease (ILD) / pneumonitis and nominal ocular toxicities (such as severe dry eye or keratitis).
Impact:
Potentially best-in-class: Achieving a 65% RRR against an established checkpoint inhibitor standard-of-care sets a new benchmark for ADC-checkpoint combinations in unselected or PD-L1-selected NSCLC populations. By delivering a cytotoxic payload directly to the tumor site with higher specificity, it bypasses traditional mechanisms of platinum-doublet or checkpoint-inhibitor resistance.
Competitive with PD1 x VEGF bsAbs: While cross-trial comparisons carry inherent limitations due to differing baseline patient characteristics and trial protocols, a look at ivonescimab’s HARMONi-2 results shows that sac-TMT demonstrated a numerically higher magnitude of benefit over Keytruda in its respective study cohort regarding relative risk reduction of progression-free survival (65% vs. 49%) and overall response rate (70% vs. 50%).


Next steps: Kelun-Biotech will leverage these registrational data to submit a Supplemental New Drug Application (sNDA) to China’s National Medical Products Administration (NMPA) for this combination. Merck & Co. (which holds the global ex-Greater China rights to MK-2870) is aggressively validating these findings globally. A massive, international Phase 3 program, including the MK-2870-004 and multi-cohort frontline studies, is actively accruing a diverse, global patient population to confirm these benefits across North American, European, and Asian centers. Future presentation readouts will dissect the data by PD-L1 expression levels (TPS <1%, 1-49%, and ≥50%) and tumor histology (squamous vs. non-squamous) to precisely identify which patient subsets derive the absolute highest magnitude of benefit from this combination.
Apogee - Zumilokibart (anti-IL13 mAb) / Phase 2 (atopic dermatitis)
Indication: Atopic Dermatitis (AD) is a chronic inflammatory skin disease driven by a complex interplay of epidermal barrier dysfunction, immune dysregulation, and microbial dysbiosis. Pathophysiologically, structural defects in the skin barrier, often arising from loss-of-function mutations in the filaggrin (FLG) gene or altered epidermal lipid composition, lead to increased transepidermal water loss and easy penetration of environmental allergens and irritants. This barrier breach prompts keratinocytes to release alarmins that selectively activate a polarized T-helper 2 (Th2) immune response, generating high levels of type 2 cytokines (primarily IL-4, IL-13, and the highly pruritogenic IL-31) that concurrently worsen the structural barrier and drive intense, non-histaminergic pruritus while promoting Staphylococcus aureus colonization. According to updated American Academy of Dermatology (AAD) guidelines, the foundational standard of care mandates frequent application of fragrance-free emollients to repair the physical barrier alongside proactive, anti-inflammatory topical therapies, such as topical corticosteroids (TCS), topical calcineurin inhibitors (tacrolimus, pimecrolimus), phosphodiesterase-4 (PDE-4) inhibitors (crisaborole, roflumilast), or topical JAK inhibitors (ruxolitinib) and aryl hydrocarbon receptor (AhR) agonists (tapinarof), to control localized flares. For moderate-to-severe disease refractory to standard topicals, management escalates directly to targeted systemic biological therapies, including the anti-IL-4R-alpha antibody dupilumab and the anti-IL-13 antibodies tralokinumab or lebrikizumab, or oral JAK inhibitors (upadacitinib, abrocitinib) to provide rapid, comprehensive systemic clearing and profound itch relief.
Mechanism: Zumilokibart is a subcutaneously administered, potential best-in-class monoclonal antibody targeting Interleukin-13 (IL-13). IL-13 is a central type 2 helper T-cell (Th2) cytokine that serves as a primary, foundational driver of the pathogenesis of atopic dermatitis. It stimulates epidermal barrier breakdown, downregulates structural proteins like filaggrin, and directly mediates severe pruritus (itch). Unlike first-generation anti-IL-13 or anti-IL-4R$\alpha$ agents, zumilokibart is specifically engineered with enhanced binding affinity to the neonatal Fc receptor (FcRn). This modification protects the antibody from intracellular degradation and recycling pathways, resulting in a significantly prolonged systemic half-life. In head-to-head preclinical assays, zumilokibart demonstrated equivalent or superior inhibition of downstream IL-13 cellular signaling cascades relative to established competitors like lebrikizumab.
Trial design: The Phase 2 APEX Part B clinical trial was a randomized, double-blind, placebo-controlled, induction and dose-optimization study that included 346 adult patients presenting with moderate-to-severe atopic dermatitis. Participants were randomized in a 1:1:1:1 ratio into four separate cohorts: High-dose zumilokibart, Mid-dose zumilokibart, Low-dose zumilokibart, or Placebo. The primary endpoint was the proportion of patients achieving an Eczema Area and Severity Index 75% improvement (EASI-75) from baseline at Week 16. Key secondary endpoints included investigator’s Global Assessment clear or almost clear skin (IGA 0/1), 90% skin clearance (EASI-90), complete clearance (EASI-100), and a ≥4-point reduction on the Itch Numeric Rating Scale (I-NRS).
Data (press release, slide deck):
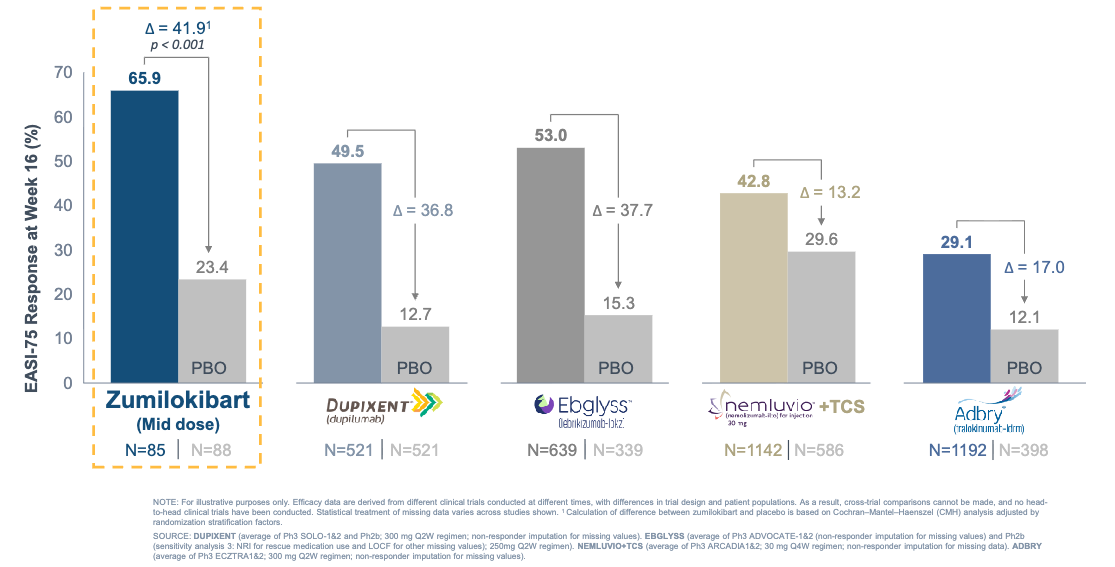
Efficacy: The trial met its primary and secondary endpoints with high statistical significance, with the mid-dose cohort emerging with the optimal therapeutic index. With the caveat that cross-trial comparisons are indirect and non-head-to-head, these 16-week EASI-75 induction rates compare favorably to historical data from pivotal monotherapy trials of currently approved biologics.
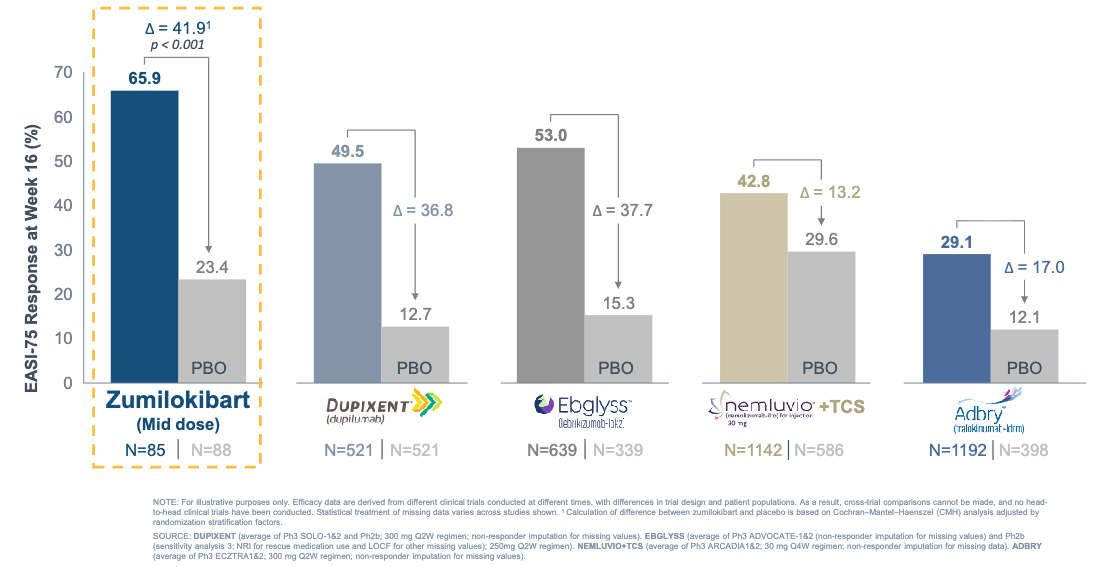
Zumilokibart demonstrated competitive EASI-75 response; Source: Apogee presentation, slide 19 Safety: Zumilokibart was well tolerated with no new or unexpected safety signals. The most common treatment-emergent adverse events (TEAEs) were nasopharyngitis, headache, and noninfective conjunctivitis. Crucially, conjunctivitis tracked in a dose-dependent manner, validating the mid-dose over the high-dose (10.6% in mid-dose vs. 20.7% in high-dose).
Impact:
Potentially best-in-class: On an informal cross-trial basis, the mid-dose 16-week induction data (EASI-75 of 65.9% and IGA 0/1 of 46.0%) compare favorably to the historical data reported in the monotherapy registration trials of standard-of-care treatments, including dupilumab (Dupixent) and lebrikizumab (Ebglyss).
Potentially Reduced the Injection Burden: Standard atopic dermatitis biologics require bi-weekly or monthly injections, which can hamper real-world compliance. Combined with APEX Part A data showing that initial responders can effectively maintain or even deepen their clinical clearance on a 3-month or 6-month maintenance schedule, zumilokibart establishes a paradigm where patients may only need 2 to 4 dosing days per year.
Next steps: Bolstered by these dose-optimization results and a fortified balance sheet, Apogee plans to initiate its Phase 3 registrational program for moderate-to-severe atopic dermatitis in the second half of 2026. The planned pivotal program will consist of two replicate monotherapy studies and one concomitant topical steroid combination study. Since IL-13 drives broader type 2 inflammatory diseases, Apogee is leveraging zumilokibart’s extended half-life across other indications. The company will launch the ELEVATE Phase 2a trial in Eosinophilic Esophagitis (EoE) in the second half of 2026 (testing 3- and 6-month dosing) and expects to advance the ASPIRE Phase 2b trial in moderate-to-severe asthma in the first half of 2027.
Arrowhead - ARO-INHBE (INHBE RNAI) / Phase 1/2a (obesity & MASH)
Arrowhead Pharmaceuticals presented updated interim Phase 1/2a data for its investigational RNA interference (RNAi) therapeutic, ARO-INHBE, at the European Association for the Study of the Liver (EASL) 2026 Congress.
Indication: We covered the obesity in a two-part Frontiers in Medicine series (Part 1, Part 2). Metabolic Dysfunction-Associated Steatohepatitis (MASH) develops when systemic metabolic dysregulation, frequently driven by obesity, insulin resistance, and type 2 diabetes, overwhelms hepatic lipid metabolism, causing an excessive accumulation of toxic lipid species (lipotoxicity) within hepatocytes. This chronic fatty infiltration triggers mitochondrial stress, endoplasmic reticulum dysfunction, and the production of reactive oxygen species, resulting in hepatocyte ballooning, lobular inflammation, and the subsequent activation of hepatic stellate cells into collagen-producing myofibroblasts that drive progressive liver fibrosis. In clinical practice, risk stratification is guided completely by non-invasive tests (NITs) such as FIB-4, vibration-controlled transient elastography (VCTE), or the Enhanced Liver Fibrosis (ELF) score rather than routine liver biopsies. According to current consensus guidelines, the foundational standard of care mandates rigorous lifestyle modification, aiming for a ≥7–10% total body weight loss via structural exercise and a Mediterranean or low-glycemic dietary framework to resolve steatohepatitis. For individuals with moderate-to-advanced, non-cirrhotic fibrosis (stages F2 and F3), clinical guidelines recommend pharmacological intervention: resmetirom, an oral thyroid hormone receptor-beta (THR-β) selective agonist that directly restores hepatic lipid homeostasis and reduces fibrosis, or semaglutide, which is preferred for treatment-naïve patients presenting with comorbid obesity and type 2 diabetes to concurrently resolve steatohepatitis and provide robust extrahepatic cardiometabolic protection.
Mechanism: ARO-INHBE leverages RNAi to target adipocyte dysfunction and liver fat accumulation via a novel, non-incretin pathway. It is designed to selectively reduce the hepatic expression of the INHBE (inhibin subunit beta E) gene. The gene product, Activin E, is a secreted signaling protein that acts as a ligand in pathways regulating energy homeostasis and fat storage in adipose tissue. Activin E natively acts as a hormonal ligand that signals the ALK7 receptor on adipose tissue to suppress lipolysis and drive fat storage. By deploying a liver-targeted siRNA to downregulate hepatic INHBE expression, ARO-INHBE safely lowers systemic Activin E. This knockdown enhances catecholamine sensitivity within fat tissue, unlocking lipolysis, reversing adipose tissue hypertrophy, and reducing visceral adiposity while protecting lean muscle mass. Human genetic studies show that loss-of-function variants in INHBE correlate with healthier fat distribution, lower visceral adiposity, and a reduced lifetime risk of type 2 diabetes. By lowering systemic Activin E levels, ARO-INHBE is designed to investigate whether enhancing lipolysis can mitigate adipose tissue hypertrophy and cellular dysfunction.
Trial design: The presented data stems from the ongoing, dose-escalating Phase 1/2a trial (NCT06700538) evaluating up to 78 adult volunteers with obesity, with and without Type 2 Diabetes Mellitus (T2DM). Part 1 (Monotherapy) evaluated safety, pharmacokinetics, and pharmacodynamics across single- and multiple-ascending doses of ARO-INHBE. Part 2 (Combination) evaluated multiple doses of ARO-INHBE administered in tandem with low-dose tirzepatide (5 mg), a dual GLP-1/GIP receptor co-agonist. The congress presentation highlighted an exploratory efficacy cohort focusing on participants with a high initial metabolic burden, defined by a baseline liver fat content (LFC) greater than 8% (mean baseline: 14.5% ± 5.1%, n=10).
Data (press release, poster):
Efficacy: A single 400 mg dose of ARO-INHBE monotherapy achieved a mean maximum reduction of 85.3% in serum Activin E. Crucially, this pharmacodynamic silencing effect persisted beyond 3 months (90 days). In the baseline liver fat content (LFC) >8% cohort, patients receiving monotherapy doses of 200 mg or greater experienced a placebo-adjusted post-dose LFC reduction of 44% (p < 0.01). In the combination cohorts (ARO-INHBE + 5 mg tirzepatide), adding the RNAi therapy resulted in significantly enhanced reductions in both visceral adipose tissue and LFC compared to treating with tirzepatide alone. Extended exposure tracking revealed that the therapeutic response was progressive, demonstrating continued, compounding improvements in visceral fat and LFC from Week 12 through Week 24.
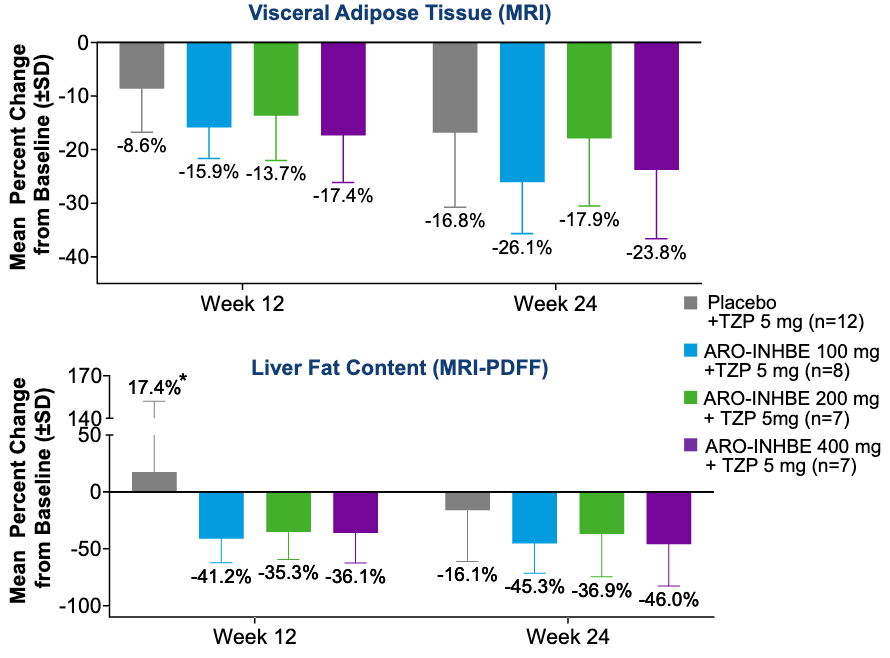
Participants with Obesity Receiving ARO-INHBE + Tirzepatide 5 mg Combination Therapy (Non-T2DM); Source: Arrowhead poster, Figure 3B & 3C Safety: ARO-INHBE was generally well tolerated as both a monotherapy and when paired with tirzepatide. The vast majority of treatment-emergent adverse events (TEAEs) were mild, with mild and self-limiting injection site reactions being the most common. Crucially, no adverse events led to study drug discontinuation or trial dropouts.
Impact:
Adding to Incretins: While standard GLP-1 and GIP therapies primarily drive weight loss via central appetite suppression, ARO-INHBE acts directly on peripheral adipose tissue biology.
A Dual Path for Obesity and MASH: The EASL data validates that silencing Activin E addresses systemic lipotoxicity. The finding that it significantly cuts liver fat content by a placebo-adjusted 44% supports the evaluation of this asset as a potential cross-over therapy designed to address obesity and reduce hepatic steatosis.
Twice-Per-Year Maintenance Potential: Because the target silencing remains durable past the 3-month mark, the data confirms that therapeutic efficacy can be maintained with an ultra-convenient, twice-yearly (bi-annual) dosing schedule.
Next steps: Backed by these liver fat clearance signals, Arrowhead leadership confirmed they are actively engaging with global regulatory authorities to map out specific clinical trial designs and primary endpoints for upcoming Phase 2 studies focusing explicitly on MASH and obesity. Investigators will continue tracking safety, durability, and body composition changes across the remaining cohorts in the AROINHBE-1001 protocol as the trial moves toward full completion.
To contact us, please send us an email at biotechreadout@gmail.com
Disclaimers
Investigational Status Disclaimer
The therapeutic candidates discussed in this newsletter are currently in clinical development and have not been approved for commercial sale by the U.S. Food and Drug Administration (FDA), the European Medicines Agency (EMA), or other global regulatory authorities. Their safety and efficacy have not been established. References to pipeline products and ongoing clinical trials involve significant risks and uncertainties. Statements regarding the potential safety, potency, or efficacy of investigational drugs reflect current hypotheses and are not a guarantee of future performance or regulatory clearance. The outcome of clinical trials is inherently unpredictable, and clinical results from earlier stages may not be predictive of results in later, larger-scale trials.
No Medical Advice Disclaimer
This newsletter is for informational and educational purposes only. The content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or another qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read in this publication.
No Patient-Provider Relationship Disclaimer
The information provided in this newsletter is for educational and analytical purposes only. Receipt of this information, or any interaction with this content, does not create a physician-patient, pharmacist-patient, or any other professional-provider relationship between you and the authors or publishers. This newsletter should not be used as a substitute for a personal consultation with a qualified healthcare professional.
Forward-Looking Statements Disclaimer
This newsletter contains “forward-looking statements” regarding future events, including clinical trial timing, regulatory milestones, and projected market performance. These statements are based on current expectations and assumptions that are subject to significant risks and uncertainties. Actual results may differ materially from those expressed or implied. We undertake no obligation to update these statements as a result of new information or future developments.
Third-Party Links & Content Disclaimer
This newsletter contains links to third-party websites, including clinical trial registries and corporate presentations. Biotech Readout does not endorse, guarantee, or assume responsibility for the accuracy or reliability of any information offered by third-party providers.
Errors and Omissions Disclaimer
While we strive for technical accuracy, the information in this newsletter is provided on an “as is” basis with no guarantees of completeness, accuracy, or timeliness. Biotech Readout assumes no liability for any errors or omissions in the content of this publication.
Non-Endorsement Disclaimer
Any reference to specific commercial products, processes, or services by trade name, trademark, or manufacturer does not constitute or imply an endorsement or recommendation by the author. All trademarks are the property of their respective owners.
No Investment Advice Disclaimer
This newsletter is for informational purposes only and does not constitute financial, investment, or legal advice. The author is not a registered investment advisor. You should consult with a professional financial advisor before making any investment decisions. The biotechnology sector is highly volatile; past performance is not indicative of future results.
Conflict of Interest Disclaimer
The author of this newsletter maintains a position of independence. At the time of publication, the author holds no direct financial interest, equity, or options in any of the companies mentioned in this report. No compensation has been received from any third party to feature or analyze specific therapeutic candidates or corporate entities.