
>30% weight loss, new blood pressure drug, FcRn cracks rheumatoid arthritis, and more
Weekly Readout #8: Week ending May 22, 2026

Disclaimer: This newsletter is for educational and informational purposes only and does not constitute medical, investment, or financial advice, nor does it establish a provider-patient relationship. Content may include forward-looking statements and discussions of investigational therapeutic candidates that are not FDA/EMA approved; their safety and efficacy remain unestablished and clinical outcomes are unpredictable. While we strive for accuracy, all information is provided as is without guarantees. This newsletter is independent, and the author holds no financial positions in the companies mentioned nor receives third-party compensation for this coverage. Please find a complete version of our disclaimers at the bottom of this article and on our About page.

Press ⬇️ to go down to a section and ⬆️ to go back up to the Table of Contents
Table of Contents
This week, we discuss…
…a newly approved medicine:
First-in-class ASI for hypertension; AstraZeneca - Baxfendy (ASI) / FDA approval (hypertension) ⬇️
…potentially first-in-class or best-in-disease treatments:
For obesity with weight loss similar to bariatric surgery; Eli Lilly - Retatrutide (GGG) / Phase 3 (obesity) ⬇️
Immunovant & Roivant - IMVT-1402 (FcRn inhibitor) / Phase 2b (D2T RA) ⬇️
Pfizer - PF-07872412 (25-valent vaccine) / Phase 2 (pneumococcal) ⬇️
Relay - RLY-2608 (PI3Kα inhibitor) / Phase 2 (Vascular Anomalies) ⬇️
Beam Therapeutics - BEAM-302 (adenine base editor) / Phase 1/2 (AATD) ⬇️
Corvus - Soquelitinib (ITK inhibitor) / Phase 1 (atopic dermatitis) ⬇️
…and other data releases:
Merck - Sacituzumab Tirumotecan (TROP2 ADC) / Phase 3 (endometrial cancer) ⬇️
Eli Lilly - Retatrutide (GGG) / Phase 3 (obesity)
Eli Lilly has announced the highly anticipated topline Phase 3 results for retatrutide from the TRIUMPH-1 trial. This data establishes retatrutide as a breakthrough in the anti-obesity pipeline, achieving unprecedented weight reduction that directly rivals outcomes from bariatric surgery.
Indication: We covered the obesity in a two-part Frontiers in Medicine series (Part 1, Part 2).
Mechanism: Retatrutide is a first-in-class, single-peptide, triple hormone receptor agonist (GIP, GLP-1, and Glucagon = GGG). It acts as a balanced “triagonist” by engaging three distinct metabolic pathways:
GLP-1 (Glucagon-Like Peptide-1): Enhances glucose-dependent insulin secretion, slows gastric emptying, and works centrally in the hypothalamus to reduce appetite and increase satiety.
GIP (Glucose-Dependent Insulinotropic Polypeptide): Acts synergistically with GLP-1 to maximize insulin sensitivity, optimize adipose tissue lipid storage, and significantly blunt the gastrointestinal side effects typically caused by pure GLP-1 agonists.
Glucagon (GCGR): The differentiating x-factor. Historically, glucagon was avoided because it elevates blood sugar. However, when balanced against GLP-1 and GIP, it does not cause hyperglycemia. Instead, it drives thermogenesis (increasing resting energy expenditure) and accelerates hepatic fat oxidation, burning lipid stores directly in the liver.
Trial design: The Phase 3 TRIUMPH-1 trial evaluated once-weekly subcutaneous injections of retatrutide (4 mg, 9 mg, or 12 mg) or a placebo in 2,339 adult participants with obesity or overweight, featuring at least one weight-related comorbidity (e.g., hypertension, dyslipidemia, cardiovascular disease), but without type 2 diabetes. All arms started at 2 mg and escalated every 4 weeks to their target dose. The primary endpoint was evaluated at 80 weeks. A pre-specified, blinded extension tracked participants with severe obesity (baseline BMI ≥35) through 104 weeks (2 years).
Data (press release):
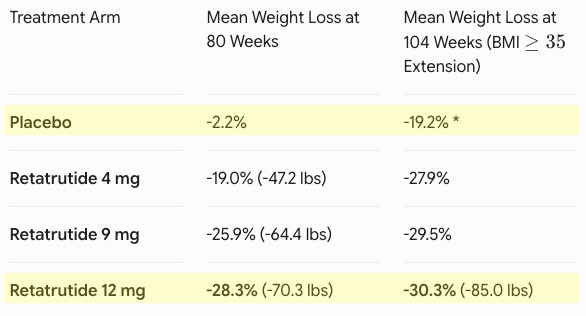
Efficacy: Retatrutide demonstrated an unprecedented, progressive weight loss trajectory (see table below). At 80 weeks, 65.3% of participants on the 12 mg dose dropped below a BMI of 30, effectively moving out of the clinical definition of obesity entirely. This included 37.5% of individuals who began the trial with Class 3 severe obesity (BMI ≥40). Marked improvements were recorded across baseline waist circumference (-24.1 cm for the 12 mg group), triglycerides, non-HDL cholesterol, systolic blood pressure, and high-sensitivity C-reactive protein (hsCRP).
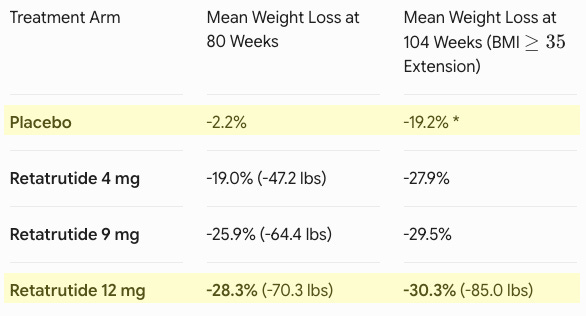
*The placebo group transitioned to the maximum tolerated dose (MTD) of retatrutide during the extension period, explaining their late-stage drop. Safety: The safety profile aligned with the established incretin class. The most frequent adverse events were gastrointestinal (nausea, diarrhea, vomiting, constipation). Notably, the 4 mg dose required only a single escalation step and demonstrated robust efficacy (19.0% at 80w) alongside lower discontinuation rates due to side effects compared to the placebo, highlighting a highly tolerable option for patient-tailored dosing.
Impact:
Potentially best-in-disease: Previously, standard-of-care options like semaglutide (Wegovy, ~15% weight loss) and tirzepatide (Zepbound, ~21-22% weight loss) vastly improved upon early weight-loss drugs but remained distinct from surgical intervention. Retatrutide’s -30.3% mean weight loss at 104 weeks represents a landmark achievement. It appears to bridge the gap with sleeve gastrectomy, which typically results in 25% to 30% total body weight loss at two years. For the first time, nearly half of the patients (45.3%) on the maximum dose achieved a level of weight reduction previously thought impossible without bariatric surgery.
Next steps: Eli Lilly plans to present the detailed, granular master data from TRIUMPH-1 at the upcoming 86th Annual American Diabetes Association (ADA) Scientific Sessions from Friday, June 5 through Monday, June 8, 2026. Results from other critical trials in the retatrutide pipeline are expected later this year, including TRIUMPH-2 (patients with type 2 diabetes) and TRIUMPH-3 (patients with established cardiovascular disease). Following the completion of the wider TRIUMPH phase 3 clinical program, Eli Lilly plans to utilize this data to anchor global regulatory filings for commercial approval.
AstraZeneca - Baxfendy (ASI) / FDA approval (hypertension)
AstraZeneca has received FDA approval for Baxfendy (baxdrostat). This approval marks the introduction of a first-in-class therapeutic option to clinical practice, targeting patients with persistently uncontrolled and treatment-resistant hypertension.
Indication: Uncontrolled hypertension generally stems from a combination of poor medication adherence, suboptimal dosing regimens, and lifestyle factors (such as high sodium intake, alcohol use, and physical inactivity) superimposed on an overactive sympathetic nervous system and progressive arterial stiffening. Resistant hypertension represents a distinct, high-risk subset defined as blood pressure that remains above target despite the concurrent use of three antihypertensive medication classes, including a blocker of the renin-angiotensin system (an ACE inhibitor or ARB), a calcium channel blocker (CCB), and a long-acting thiazide-like diuretic. Its pathophysiology is heavily driven by chronic Aldosterone-mediated fluid and sodium retention, coupled with heightened sympathetic drive and secondary causes like obstructive sleep apnea or renal artery stenosis. Consequently, while the broader standard of care for uncontrolled hypertension focuses on maximizing adherence and optimizing standard first-line therapies, the foundational guideline-directed management for true resistant hypertension necessitates adding a mineralocorticoid receptor antagonist (MRA) like spironolactone as the essential fourth-line agent, alongside targeted treatment of any underlying secondary causes.
Mechanism: Baxfendy is a highly selective and potent small-molecule aldosterone synthase inhibitor (ASI). It works downstream of the renin-angiotensin-aldosterone system (RAAS) to lower blood pressure through a novel pathway. It selectively targets CYP11B2 (aldosterone synthase), the foundational enzyme responsible for synthesis of aldosterone in the adrenal cortex. While standard drugs like ACE inhibitors or ARBs block upstream components, chronic therapy often triggers “aldosterone escape,” where aldosterone levels rebound and cause fluid/sodium retention, arterial stiffening, and fibrosis. Baxfendy stops aldosterone production at its absolute source. Crucially, Baxfendy possesses high selectivity for aldosterone synthase over CYP11B1 (cortisol synthase), allowing it to drastically reduce circulating aldosterone without suppressing the body’s natural cortisol production.
Trial design (label, NEJM paper, press release): The registrational BaxHTN Phase 3 once-weekly/daily oral doses of Baxfendy (1 mg or 2 mg) or a matching placebo on top of standard-of-care medications in 796 adult participants with confirmed uncontrolled or treatment-resistant hypertension. All patients had persistently high blood pressure despite taking a stable background regimen of at least two or more standard antihypertensive medications (e.g., ACEi/ARBs, CCBs, or diuretics). The primary efficacy endpoint evaluated the absolute and placebo-adjusted change in mean seated systolic blood pressure (SBP) from baseline to Week 12.
Data:
Efficacy: At Week 12, both Baxfendy 1 mg and 2 mg were superior to placebo in reducing systolic blood pressure and diastolic blood pressure. The mean change from baseline in systolic blood pressure over time in each treatment arm is shown in the graph below. The treatment benefit of Baxfendy over placebo on the primary endpoints was generally consistent across pre-specified subgroups, including age, sex, BMI, renal function (eGFR), and systolic blood pressure at baseline.
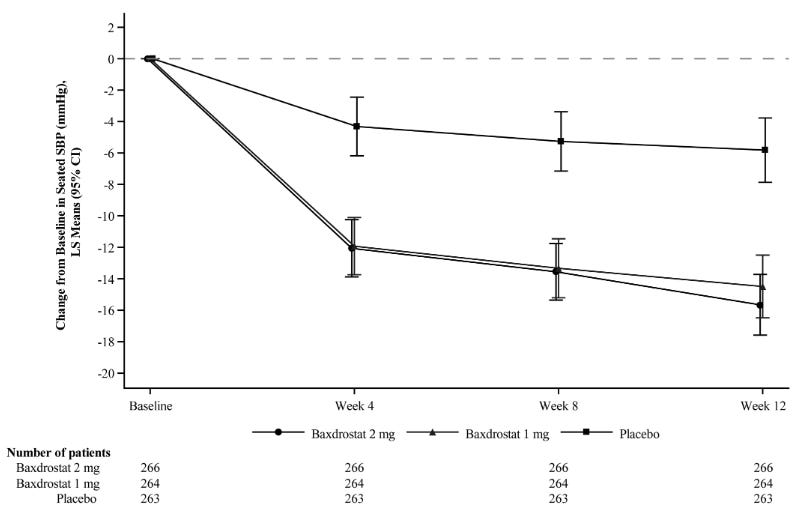
Mean and 95% Confidence Intervals for Change from Baseline in Seated Systolic Blood Pressure (SBP) Over 12 Weeks, BaxHTN Trial; Source: FDA label for Baxfendy Safety: Baxfendy was well-tolerated with no unexpected adverse safety signals. The most frequent side effects observed were hyperkalemia, hyponatremia, hypotension, dizziness, and muscle spasms. The drug’s official FDA label notes no contraindications. Dosing is lower for patients with electrolyte imbalances (e.g., elderly ≥65 years old, or those with chronic kidney disease/diabetes).
Impact:
First-in-class ASI: In a therapeutic market saturated with historical, decades-old medication classes, Baxfendy represents a new option for patients with uncontrolled and resistant hypertension. Approximately 50% of the 1.4 billion people globally with hypertension remain uncontrolled despite taking multiple drugs.
Next steps: AstraZeneca is initiating the commercial rollout of Baxfendy tablets (available in 1 mg and 2 mg strengths) to pharmacies immediately. Seeking to expand Baxfendy beyond standalone refractory hypertension, AstraZeneca is actively enrolling a Phase III trial investigating a combination product of baxdrostat + dapagliflozin (Farxiga; SGLT2 inhibitor). This trial evaluates cardiovascular and renal survival outcomes in patients suffering from both chronic kidney disease (CKD) and high blood pressure. Armed with the FDA approval, AstraZeneca is preparing data packages to secure marketing authorization across the European Medicines Agency (EMA), Japan’s PMDA, and other major international regulatory bodies.
Immunovant & Roivant - IMVT-1402 (FcRn inhibitor) / Phase 2b (D2T RA)
Immunovant and Roivant Sciences have announced encouraging preliminary Phase 2b data for IMVT-1402, their next-generation FcRn inhibitor, in difficult-to-treat rheumatoid arthritis (D2T RA). Following the strategic discontinuation of their first-generation asset batoclimab, these results position IMVT-1402 as a potential blockbuster capable of breaking through historical efficacy ceilings in highly refractory autoimmune populations.
Indication: The pathophysiology of difficult-to-treat rheumatoid arthritis (D2T RA) involves a highly complex, heterogeneous interplay of persistent synovial inflammation, alternative cytokine signaling (e.g., shifts between TNF, IL-6, and JAK/STAT pathways), and structural joint damage that remains active despite conventional interventions. This resistance is frequently amplified by non-inflammatory factors, including structural joint damage mimicking active inflammation, secondary osteoarthritis, fibromyalgia, and altered central pain processing. Consequently, the European Alliance of Associations for Rheumatology (EULAR) standard of care shifts away from standard cycling of traditional biologic or targeted synthetic disease-modifying antirheumatic drugs (b/tsDMARDs). Instead, the clinical framework mandates a comprehensive re-evaluation to confirm true inflammatory activity versus non-inflammatory pain, systematically address comorbidities and poor adherence, and utilize a multidisciplinary approach combining non-pharmacological modalities, psychological support, and personalized, sequential switching of advanced therapies targeted to the patient’s dominant disease phenotype.
Mechanism: IMVT-1402 is a fully human, next-generation monoclonal antibody designed to selectively inhibit the neonatal Fc receptor (FcRn). In a large subset of RA patients, the disease is driven by autoantibodies like anti-citrullinated protein antibodies (ACPA) and rheumatoid factor (RF). FcRn acts as a recycling mechanism that prevents antibodies from being destroyed. By blocking FcRn, IMVT-1402 accelerates the degradation of all Immunoglobulin G (IgG) antibodies, rapidly clearing these pathogenic autoantibodies from circulation. First-generation FcRn inhibitors (including batoclimab) caused off-target reductions in serum albumin and triggered a rise in low-density lipoprotein (LDL) cholesterol. IMVT-1402 features a unique binding profile that selectively clears IgG without disrupting albumin or lipid levels, vastly improving its safety and chronic commercial viability.
Trial design: The Phase 2b D2T RA study included 170 adult participants with definite, active ACPA-positive rheumatoid arthritis classified as difficult-to-treat (D2T). This represents a highly refractory, long-duration population with high unmet needs—all patients had failed at least two or more advanced biologic/targeted synthetic DMARDs. A major key subgroup (N=107) consisted of patients who had specifically failed both a TNF inhibitor (TNFi) and a JAK inhibitor (JAKi). During the evaluated open-label period, patients received a 600 mg weekly subcutaneous (SC) dose of IMVT-1402. To minimize bias, independent and blinded joint assessors were utilized. The interim data readout focused on clinical improvements and response rates evaluated at Week 16.
Data (Immunovant press release, Roivant press release, slide deck):
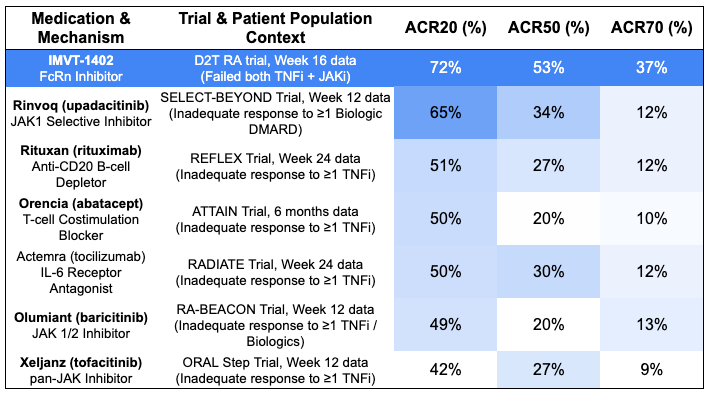
Efficacy: IMVT-1402 demonstrated profound efficacy at Week 16, dramatically outperforming historical standard-of-care (SOC) benchmarks for refractory RA patients switching to alternative biologics*.
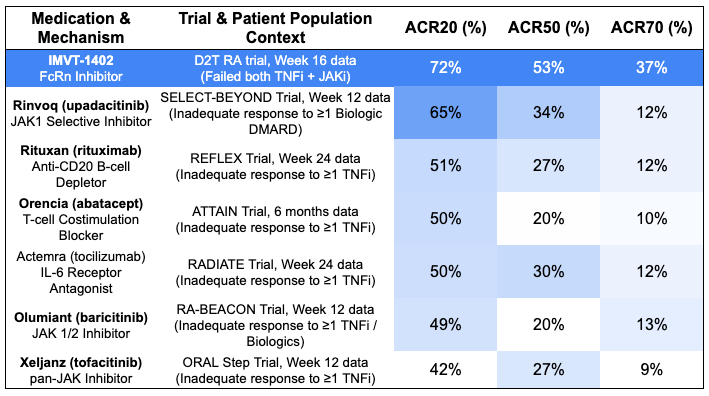
*Note: cross-trial comparisons do not account for differences in baseline characteristics, dosing schemes, and treatment durations. Safety: IMVT-1402 was “safe and well-tolerated” according to the trial sponsor, with no new drug-related safety signals identified. Crucially, confirming its next-generation design, the asset maintained excellent systemic tolerability without the albumin drop or LDL cholesterol spikes that plagued earlier-generation class competitors.
Impact:
Potentially first-in-class mechanism that overcomes mechanism exhaustion: In highly refractory TNF-failed populations, standard-of-care FDA-approved biologics (like rituximab or abatacept) and JAK inhibitors (like baricitinib or upadacitinib) historically hit a rigid therapeutic ceiling for profound clinical response, consistently maxing out at 10% to 13% for ACR70. When patients fail both a TNF blocker and a JAK inhibitor, cycling to another agent in those families usually results in diminishing returns. Because IMVT-1402 uses an entirely distinct mechanism, systemic clearing of pathogenic IgG autoantibodies via FcRn block rather than broad cytokine suppression, it circumvents the intracellular and downstream signaling exhaustion seen with traditional standard of care. IMVT-1402’s 37.4% ACR70 in an even harder-to-treat population (TNFi + JAKi failures) nearly triples this historical benchmark on a cross-trial basis.
Next steps: Immunovant plans to present more detailed, granular data from this Phase 2b trial in the second half of calendar year 2026. The company is capitalizing on its full pivot away from batoclimab to fast-track IMVT-1402. They expect to receive crucial FDA feedback regarding accelerated approval pathways for their broader portfolio by late 2026. Near-term catalysts for the asset include a Phase 2 proof-of-concept readout in Cutaneous Lupus Erythematosus (CLE) in H2 2026, followed by topline data from potentially registrational trials in Graves’ disease and Myasthenia Gravis slated for 2027.
Pfizer - PF-07872412 (25-valent vaccine) / Phase 2 (pneumococcal)
Pfizer has detailed the highly encouraging Phase 2 data for PF-07872412 (25vPnC), its fourth-generation, investigational 25-valent pneumococcal conjugate vaccine, at the International Society of Pneumonia & Pneumococcal Diseases (ISPPD) congress. The data establishes a clear evolutionary leap over Pfizer’s current market-leading vaccine, Prevnar 20, by expanding strain coverage and correcting a historical class weakness regarding a highly virulent serotype.
Indication: The pathophysiology of pneumococcal disease begins with the asymptomatic colonization of the human nasopharynx by Streptococcus pneumoniae (a encapsulated, gram-positive diplococcus). From this reservoir, the bacteria can aspirate into the lower respiratory tract or penetrate the mucosal epithelium, utilizing a thick polysaccharide capsule that inhibits host complement binding and evades phagocytosis. Once invasive, cell wall components and toxins like pneumolysin trigger a potent, dysregulated host inflammatory response and cytokine release, resulting in alveolar consolidation (pneumonia), direct invasion of the bloodstream (bacteremia), or crossing of the blood-brain barrier (meningitis). The definitive standard of care relies on prompt, empiric broad-spectrum antibiotic therapy—tailored by disease severity and regional susceptibility—typically utilizing beta-lactams (e.g., amoxicillin, ceftriaxone) often paired with a macrolide or a respiratory fluoroquinolone to address community-acquired pneumonia, alongside aggressive supportive care and adjunctive corticosteroids in cases of meningitis to blunt the damaging inflammatory response.
Mechanism: PF-07872412 is a multivalent pneumococcal conjugate vaccine designed to elicit functional, serotype-specific anti-capsular antibody responses against Streptococcus pneumoniae. It utilizes Prevnar 20 (PCV20) as its foundational backbone, carrying forward protection against 20 baseline serotypes and adding 5 new epidemiologically important serotypes: 15A, 23A, 23B, 24F, and 35B. Serotype 3 remains an aggressive, highly virulent driver of invasive pneumococcal disease (IPD) and necrotizing pneumonia in infants. Because its thick, slimy polysaccharide capsule sheds easily into surrounding tissue, traditional conjugate designs often fail to generate strong immune memory against it. PF-07872412 integrates a proprietary, next-generation carrier-protein conjugation chemistry specifically engineered to stabilize the presentation of the Serotype 3 antigen, maximizing T-cell dependent immune responses.
Trial design: The Phase 2 Infant Study was a randomized clinical trial evaluating healthy infants (enrolled starting at approximately 2 months of age). Participants were randomized to receive a standard pediatric 4-dose series of either PF-07872412 or Prevnar 20. The injections were administered concurrently with regular childhood vaccinations at 2, 4, 6, and 12–15 months of age. Primary outcomes focused on safety, tolerability, and the geometric mean titers (GMTs) of serotype-specific IgG antibodies measured exactly one month after Dose 3 (the primary infant series) and one month after Dose 4 (the toddler booster).
Data (press release):
Efficacy: While the vaccine demonstrated robust immunogenicity across all 25 targeted serotypes, the data revealed an exceptionally stark, statistically significant, multi-fold expansion in antibody levels against the historically elusive Serotype 3. The geometric mean titer (GMT) for Serotype 3 was 8.8-fold higher in the PF-07872412 cohort compared to the Prevnar 20 cohort (4.22 versus 0.48) one Month post-dose 3 (infant series). The gap widened further. The GMT for Serotype 3 was approximately 15-fold higher with PF-07872412 than with Prevnar 20 (13.85 versus 0.92) one month post-dose 4 (toddler booster). The company indicated that the strong immune responses across the other 20 matched serotypes leave them highly confident that standard regulatory non-inferiority thresholds will be met or exceeded.
Safety: The safety profile of PF-07872412 was highly consistent with currently approved, standard-of-care pediatric pneumococcal vaccines. There were no new or unexpected safety signals identified. The most frequent adverse events were transient, mild-to-moderate local injection-site reactions (redness, swelling, or pain).
Impact:
Closing the “Residual Disease” Gap: Pfizer’s Prevnar franchise (Prevnar 13 and Prevnar 20) generated nearly $6.5 billion last year (2025), commanding massive global market share. However, even with widespread adoption, Serotype 3 continues to evade complete vaccine suppression, driving up to 15% of all residual invasive pneumococcal disease in children under five. By delivering a 15-fold increase in functional antibody titers against this specific strain while simultaneously adding five new emerging serotypes, PF-07872412 expands overall serotype coverage to approximately 90% of all disease-causing strains in US children. This effectively leapfrogs competing pediatric designs by neutralizing the most dangerous remaining epidemiologic blindspots in infant preventive care.
Next steps: Armed with this Phase 2 success and regulatory alignment, Pfizer has officially initiated its global Phase 3 pediatric program in May 2026. The pivotal trial will enroll up to 2,400 healthy infants to compare the safety, tolerability, and immunogenicity of the full 4-dose series of PF-07872412 directly against standard Prevnar 20. In a bid to defend and insulate its adult vaccine market against fierce upcoming competition (such as Merck’s 21-valent adult alternative), Pfizer announced it is bypassing a 25-valent adult timeline entirely. Instead, Pfizer is moving directly to a fifth-generation, 35-valent adult pneumococcal vaccine candidate utilizing the same next-gen technology. This 35-valent adult program is slated to enter Phase 1 clinical development by the end of 2026.
Relay - RLY-2608 (PI3Kα inhibitor) / Phase 2 (Vascular Anomalies)
Relay Therapeutics has announced outstanding preliminary Phase 2 data for RLY-2608, its highly selective, pan-mutant PI3Kα inhibitor, for the treatment of PIK3CA-related vascular malformations (VM). These results demonstrate that a mutation-selective approach can deliver vastly superior, faster lesion shrinkage than currently approved non-selective alternatives.
Indication: The pathophysiology of vascular anomalies centers on a fundamental division between vascular tumors (cellular proliferations) and vascular malformations (structural defects). Vascular tumors, most notably infantile hemangiomas, are driven by localized endothelial cell proliferation and altered angiogenesis mediated by growth factors like VEGF and bFGF. Conversely, vascular malformations (classified by fluid dynamics into low-flow venous, lymphatic, or capillary, and high-flow arteriovenous malformations) are non-proliferative, structural lesions driven by somatic, post-zygotic “mosaic” mutations in critical endothelial signaling pathways—most frequently involving the PI3K/AKT/mTOR pathway (e.g., PIK3CA in PROS/CLOVES) or the RAS/MAPK pathway (e.g., MAP2K1 or RASA1). These mutations cause progressive, uncoordinated vessel enlargement, localized tissue hypertrophy, and severe pain. The modern standard of care has transitioned from historically high-risk surgical debulking and embolization to targeted precision pharmacotherapy: infantile hemangiomas are managed primarily with oral propranolol (a non-selective beta-blocker that induces endothelial apoptosis and vasoconstriction), while complex, refractory low-flow vascular malformations are increasingly treated with sirolimus (rapamycin), an oral mTOR inhibitor that directly dampens the overactive intracellular signaling driving abnormal vessel growth.
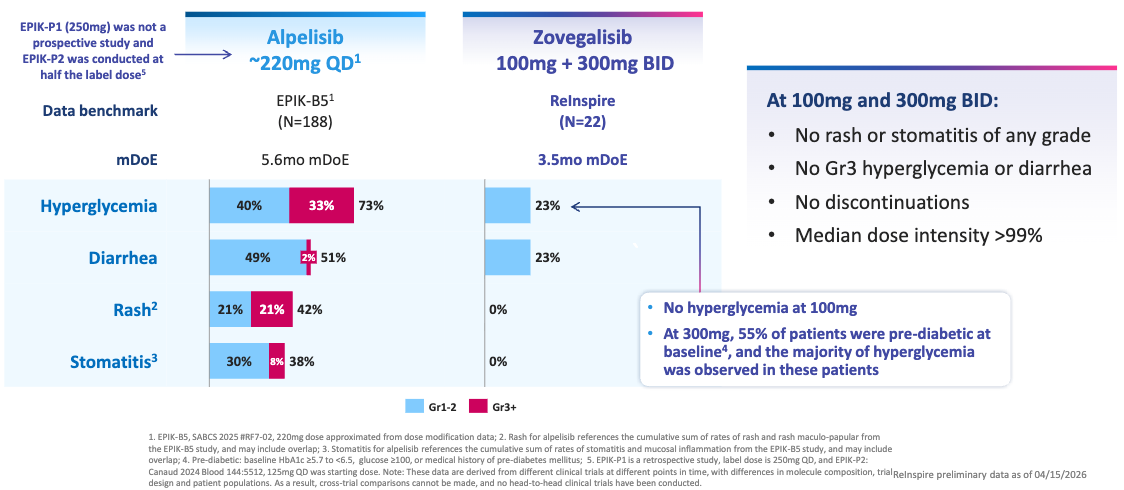
Mechanism: Vascular malformations within the PIK3CA-related overgrowth spectrum (PROS) are driven by somatic, activating mutations in the PIK3CA gene, which encodes the p110α catalytic subunit of PI3K. This causes chronic hyperactivation of the downstream AKT/mTOR pathway, leading to abnormal vessel proliferation. First-generation treatments, such as Novartis’s Vijoice (alpelisib), are orthosteric inhibitors that block both mutated and wild-type (normal) PI3Kα. Because wild-type PI3Kα is essential for systemic glucose metabolism, blocking it triggers severe, dose-limiting hyperglycemia and hyperinsulinemia. RLY-2608 is an allosteric, pan-mutant selective PI3Kα inhibitor. It preferentially binds to the altered conformation of mutated PI3Kα while sparing the wild-type enzyme. This allows for near-complete suppression of the overactive signaling inside the malformation while avoiding the metabolic side effects that limit the dosing and efficacy of older therapies.
Trial design: The Phase 2 trial evaluated a continuous, optimized daily oral dose of RLY-2608 as a monotherapy in adult and pediatric patients with severe, progressive, or highly symptomatic low-flow vascular malformations (venous, lymphatic, or capillary) confirmed to harbor a somatic PIK3CA mutation. The primary efficacy endpoint was the 12-week volumetric response rate, defined as the proportion of patients achieving a ≥20% or greater reduction in target lesion volume from baseline, as measured by independent, blinded central MRI review. Secondary endpoints included safety, pain reduction, and quality-of-life scores.
Data (press release, slide deck):
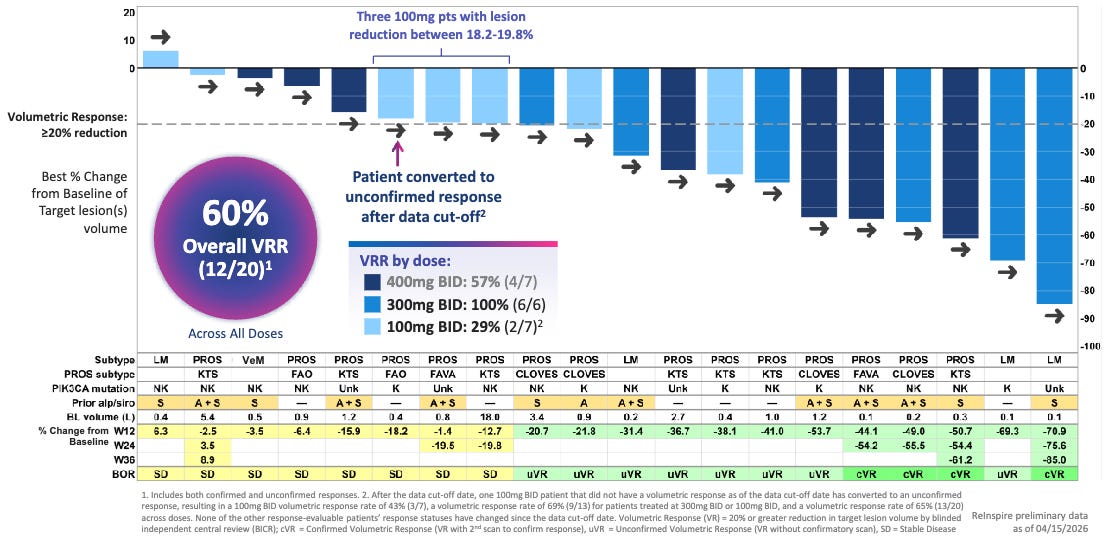
Efficacy: RLY-2608 demonstrated rapid and profound tissue cytoreduction, outpacing historical clinical benchmarks on a cross-trial basis. Approximately 60.0% of patients achieved a ≥20% target lesion volume reduction at just 12 weeks of therapy versus 37.5% volumetric response rate (≥20% reduction) with 24 weeks of Vijoice (alpelisib) in the EPIK-P1 registrational trial.
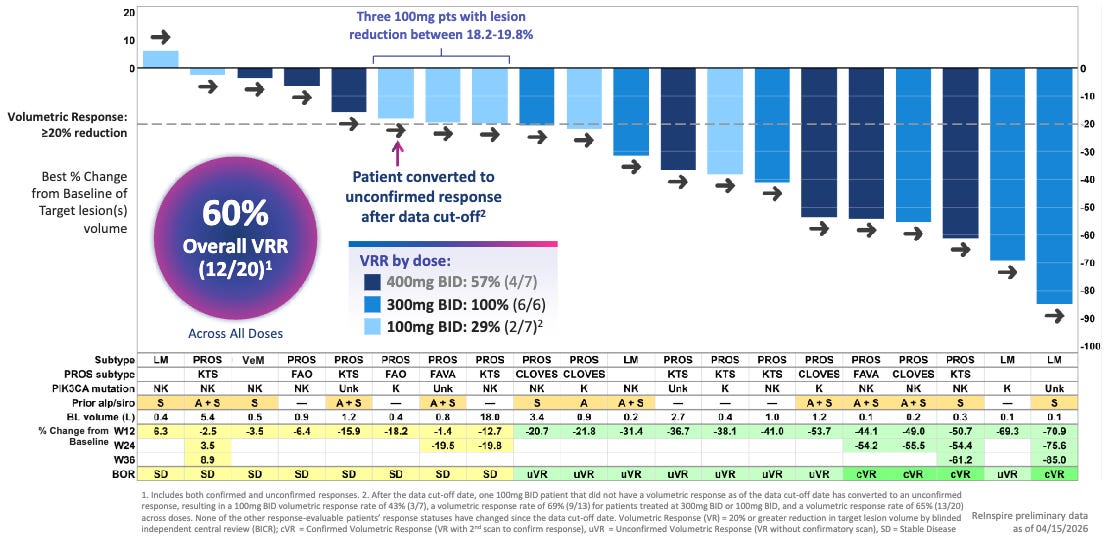
60% Volumetric Response Rate by Blinded Independent Central Review (BICR) Across All Doses; Source: Relay presentation, slide 12 Safety: Reflecting its mutant-selective mechanism, RLY-2608 showcased “a safety profile that supports chronic”, according to the trial sponsor. Unlike standard PI3Kα inhibitors, there were no reports of severe (Grade 3/4) hyperglycemia or rash, which are the leading causes of dose reductions and treatment discontinuations with Vijoice. The most common adverse events were low-grade, manageable GI upset and fatigue.
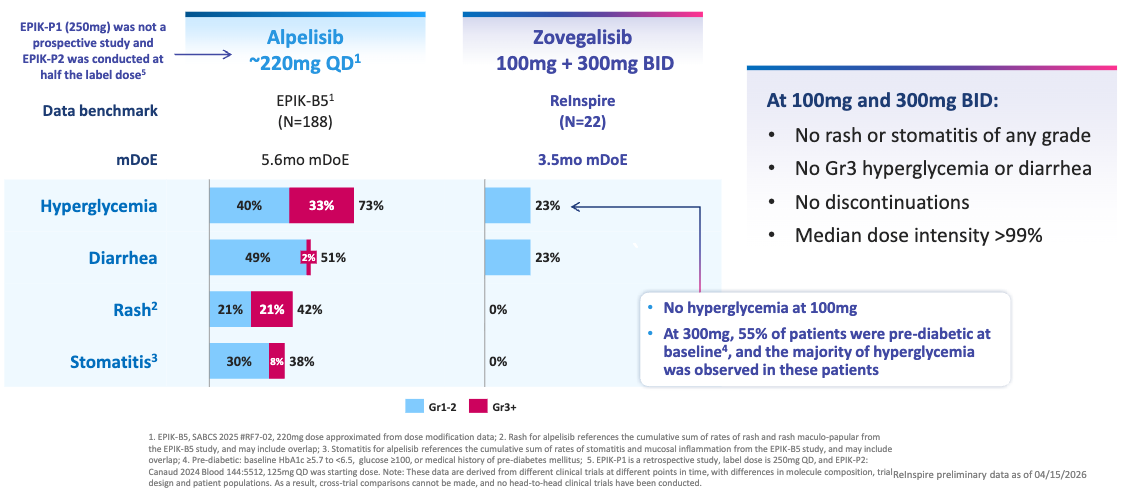
Tolerability Profile Across Known Key Pathway Adverse Events (AEs); Source: Relay presentation, slide 17 Impact:
Potentially best-in-class: For patients living with severe vascular malformations, these lesions cause debilitating pain, recurrent bleeding, functional impairment, and profound cosmetic disfigurement. While the approval of Vijoice proved the validity of targeting this pathway, its real-world utility has been hampered by modest absolute shrinkage rates and tough metabolic side effects. By hitting a 60% response rate in half the time (12 weeks vs. 24 weeks), RLY-2608 sets a new therapeutic standard. Sparing wild-type PI3Kα allows patients to stay on a fully therapeutic, uninterrupted dose, maximizing the physical collapse of the malformation and dramatically improving pain and mobility without disrupting glucose homeostasis.
Next steps: Relay Therapeutics plans to present the detailed Phase 2 data package, including pediatric cohorts and secondary patient-reported outcomes, at a major medical congress in late 2026. Relay is initiating discussions with the FDA and EMA to align on a design for a global, pivotal Phase 3 trial. Given the high unmet need and breakthrough potential, the company will likely pursue an accelerated regulatory pathway. Beyond vascular anomalies, Relay is actively exploring RLY-2608’s primary pipeline indication in PIK3CA-mutated advanced breast cancer, where data from ongoing combination trials (with endocrine and CDK4/6 therapies) are expected to read out over the coming quarters.
Beam Therapeutics - BEAM-302 (adenine base editor) / Phase 1/2 (AATD)
Beam Therapeutics presented updated Phase 1/2 clinical data for BEAM-302 at the American Thoracic Society (ATS) International Conference. The readout marks a historic milestone as the first-ever clinical in vivo base editing correction of a human genetic mutation, completely eclipsing historical endpoints and establishing a lead over RNA-editing competitors like Wave Life Sciences (WVE-006) in a cross-trial comparison.
Indication: The pathophysiology of Alpha-1 Antitrypsin Deficiency (AATD) represents a dual-organ mechanism driven by a genetic mutation in the SERPINA1 gene (most commonly the homozygous Pi*ZZ genotype), which creates a loss-of-function in the lungs alongside a toxic gain-of-function in the liver. Under normal conditions, the liver synthesizes alpha-1 antitrypsin (AAT) to act as a crucial protease inhibitor that neutralizes neutrophil elastase in the lower respiratory tract. In severe AATD, the mutant Z-AAT protein misfolds and polymerizes within the endoplasmic reticulum of hepatocytes, preventing its secretion; this intracellular accumulation triggers hepatocyte death, inflammation, and progressive liver fibrosis or cirrhosis. Concurrently, the severe drop in circulating blood levels creates an uninhibited protease environment in the lungs, where unchecked neutrophil elastase destroys alveolar elastin, leading to premature, panacinar emphysema. The historical standard of care operates on two distinct pathways: pulmonary disease is managed with standard COPD guidelines (bronchodilators, inhaled corticosteroids, and strict smoking cessation) supplemented by intravenous augmentation therapy (weekly infusions of purified human donor-derived AAT) to slow the decline of lung function, whereas hepatic disease is managed purely through supportive cirrhosis protocols and lifestyle adjustments (avoiding alcohol), as augmentation does not fix liver retention, leaving liver transplantation as the only curative option for end-stage hepatic failure.
Mechanism: BEAM-302 is an in vivo genetic medicine delivered via liver-targeting lipid nanoparticles (LNPs). It is specifically engineered to fix the homozygous Pi*ZZ mutation responsible for severe Alpha-1 Antitrypsin Deficiency (AATD). Unlike traditional CRISPR systems that cut the double-stranded DNA backbone (risking chromosomal deletions or insertions), BEAM-302 utilizes an Adenine Base Editor (ABE). It executes a seamless, chemically precise A-to-G transition directly at the PiZ mutation site on the SERPINA1 gene. By converting the mutated PiZ allele into a normal, corrected PiM allele, BEAM-302 addresses the dual pathophysiology of AATD. It simultaneously shuts down production of the toxic, misfolded Z-AAT protein (resolving liver accumulation/fibrosis) and restores systemic production of healthy, functional M-AAT protein (protecting the lungs from unchecked neutrophil elastase degradation).
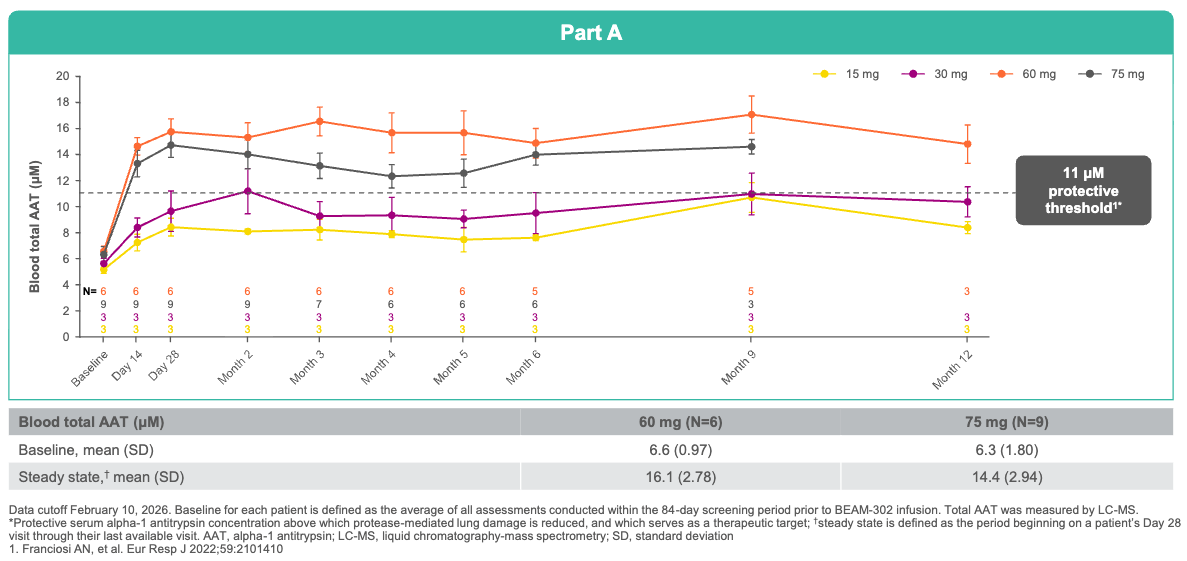
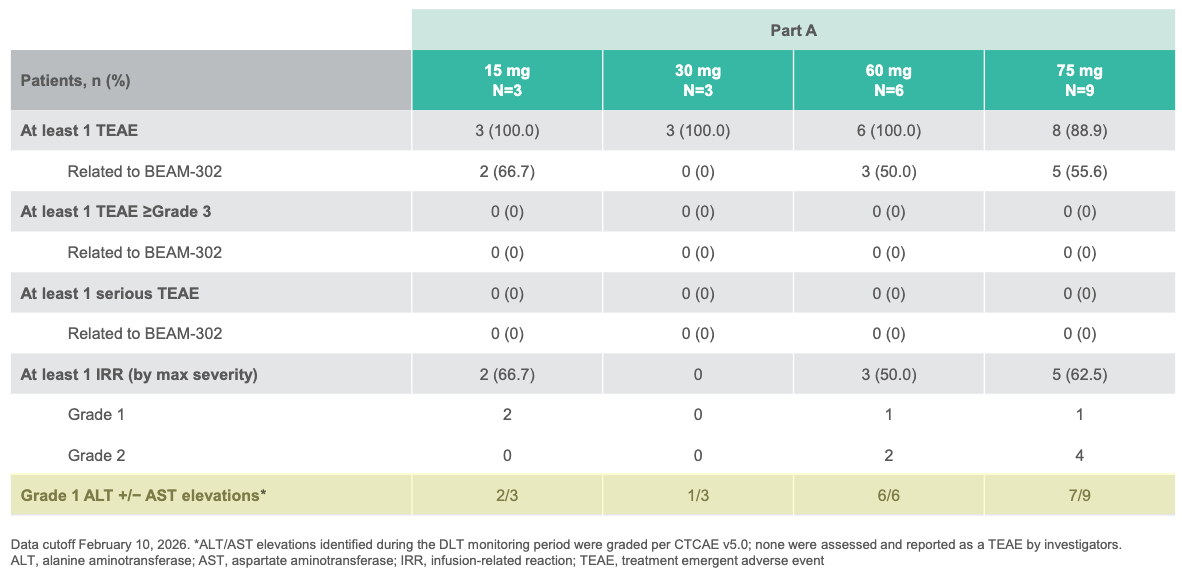
Trial design: The open-label Phase 1/2 trial evaluated a single intravenous infusions of BEAM-302 across varying cohorts (including a 60 mg and 75 mg arm), alongside a small multi-dose cohort in 29 adult patients with severe AATD harboring the homozygous PiZZ genotype, presenting with AATD-associated lung and/or liver disease. Primary metrics tracked safety, tolerability, absolute reduction of mutant Z-AAT, and the percentage/durability of newly synthesized, healthy M-AAT in circulation through 12 months of follow-up.
Data (press release, slide deck):
Efficacy: Following a single dose of BEAM-302 (60 mg), newly corrected, healthy M-AAT comprised a 91% to 94% of all total circulating AAT at steady state. For perspective, this drastically outperforms Wave Life Sciences’ RNA-editing candidate (WVE-006), which reached a maximum of 58.7% M-AAT composition after a single dose (and 70.5% after multiple doses). BEAM-302 triggered a durable 84% reduction in toxic mutant Z-AAT protein, cutting off liver injury at the source. Treated patients achieved a mean steady-state total AAT level of 16.1 µM, comfortably pushing 100% of the 60 mg cohort well above the clinical protective threshold (11 µM) needed to halt lung disease. Because BEAM-302 permanently corrects the gene in its native locus rather than inserting a foreign transgene, the corrected gene responds normally to immune triggers. During a transient respiratory infection at Month 8, a treated patient’s total AAT physiologically surged from 15.9 µM to 29.5 µM while maintaining a 95% M-AAT healthy composition, providing real-time, on-demand lung protection.
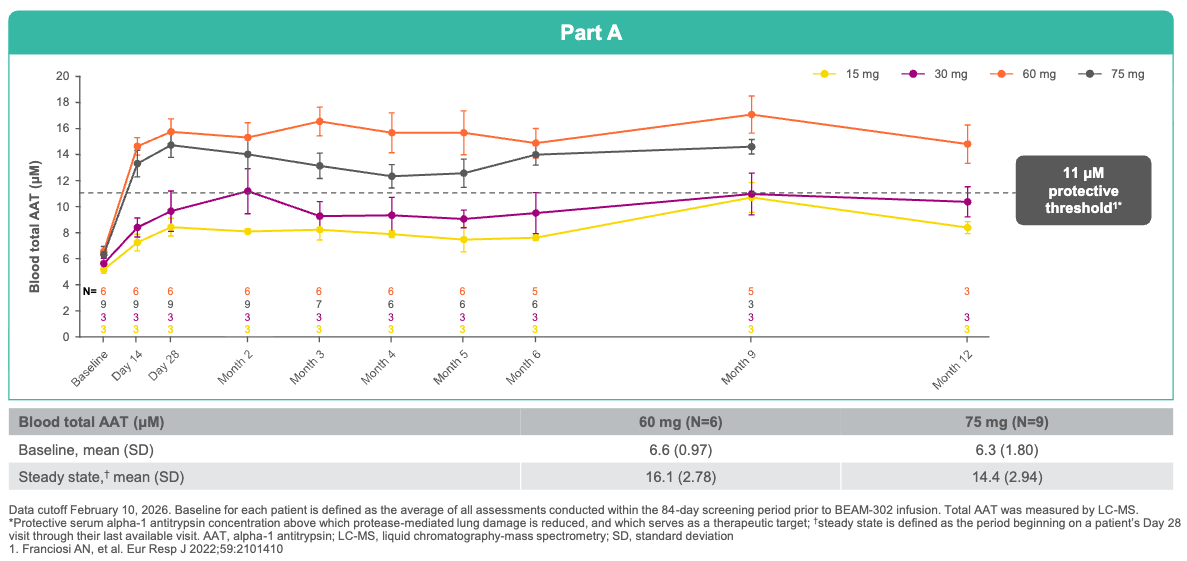
A single dose of BEAM-302 (≥60 mg) led to sustained total AAT levels above the protective threshold (≥11 μM); Source: Beam presentation, slide 12 Safety: Transient, low-grade liver enzyme elevations (ALT/AST) were observed in a couple of patients, but these were completely asymptomatic, resolved spontaneously, and required no medical intervention.
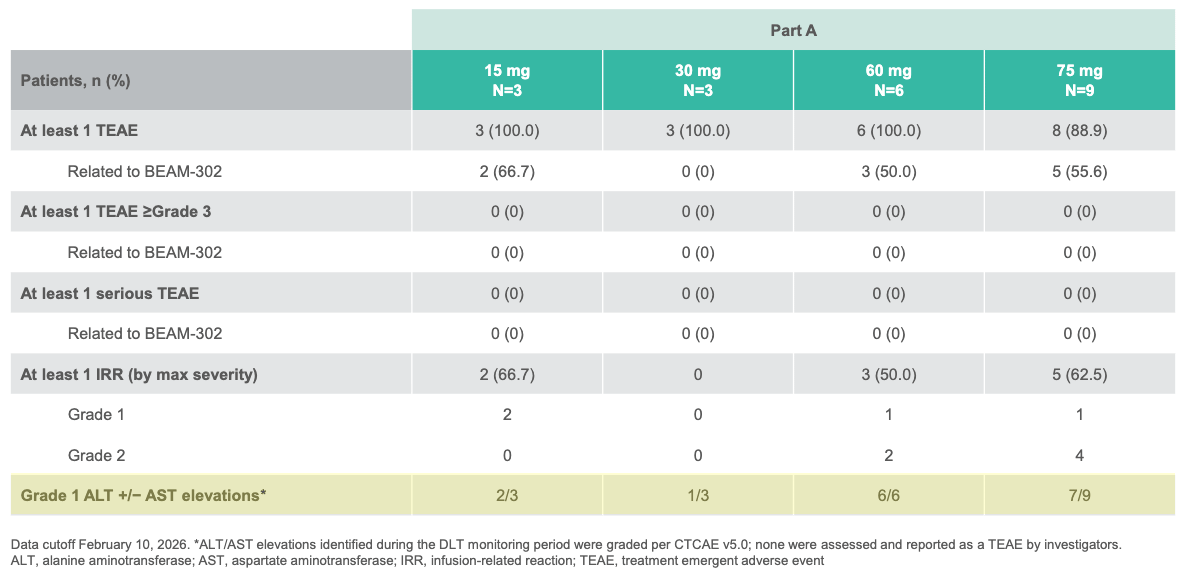
No serious adverse events or dose-dependent safety trends were observed in BEAM-302 single-dose cohorts; Source: Beam presentation, slide 11 Impact:
Potentially first- and best-in-class: The data effectively confirms that a one-time in vivo base editing infusion can permanently transition a severe, life-threatening “PiZZ” patient into a phenotype mimicking a benign “MZ” carrier (individuals who carry one normal gene, produce sufficient M-AAT, and generally live normal lifespans free of lung and liver disease). This represents a shift from historical lifelong weekly IV augmentation infusions, potentially offering a long-acting treatment for both the lungs and the liver.
Next steps: Beam has selected 60 mg as its optimal biological dose and is on track to initiate a global, pivotal phase 3 expansion cohort in the second half of 2026, enrolling roughly 50 additional AATD patients with lung or liver manifestations. Beam has successfully aligned with the FDA on a potential accelerated approval pathway utilizing these 12-month AAT biomarker endpoints as surrogate outcomes. The FDA has accepted BEAM-302 into its Chemistry, Manufacturing, and Controls Development and Readiness Pilot (CDRP) program, heavily expediting the manufacturing validation steps required for an eventual Biologics License Application (BLA).
Corvus - Soquelitinib (ITK inhibitor) / Phase 1 (atopic dermatitis)
Corvus presented the final data from this Phase 1 trial just recently at the Society for Investigative Dermatology (SID) Annual Meeting in Chicago (held May 13–16, 2026). The trial results were featured in two separate oral sessions at the conference: an initial presentation on May 14, 2026 by Dr. Kavita Sarin from Stanford University on the primary clinical and immunologic data, and a late-breaking oral presentation on May 16, 2026 by Dr. Albert Chiou from Stanford University on the prolonged, drug-free remissions seen in the Cohort 4 patients during their post-treatment observation window.
Indication: The pathophysiology of atopic dermatitis (AD) is driven by a complex interplay of genetic epidermal barrier disruption, severe immune dysregulation, and alterations in the skin microbiome. Mutations in structural genes like filaggrin (FLG) lead to a leaky, compromised skin barrier that increases transepidermal water loss and allows easy penetration of environmental allergens and irritants. This barrier breach triggers an exaggerated, Th2-mediated helper T-cell immune response dominated by the overexpression of type 2 cytokines—specifically interleukin-4 (IL-4) and interleukin-13 (IL-13)—which directly suppress key barrier proteins, activate pruriceptive sensory neurons to cause intense itching, and promote colonization by Staphylococcus aureus, further driving chronic, eczematous skin inflammation. The modern standard of care operates on a stepped approach: foundational maintenance relies on daily application of non-prescription emollients and avoidance of environmental triggers to repair the skin barrier. For mild-to-moderate flares, frontline treatments include topical anti-inflammatory agents like topical corticosteroids (TCS), topical calcineurin inhibitors (TCI, such as tacrolimus), or newer non-steroidal topicals like PDE4 inhibitors (crisaborole) and JAK inhibitors (ruxolitinib). For patients with severe, chronic, or
Mechanism: Soquelitinib is a first-in-class, oral, small-molecule selective inhibitor of IL-2-inducible T-cell kinase (ITK). It works through a unique “immune rebalancing” strategy rather than broad immunosuppression. ITK is a signaling molecule essential for T-cell differentiation and cytokine production. By selectively inhibiting ITK, soquelitinib dampens the overactivation of Th2 and Th17 pathways, blocking the downstream release of key pathogenic cytokines (IL-4, IL-5, IL-13, IL-17, and the pruriceptive cytokine IL-31). Instead of traditional blockade, ITK inhibition promotes the generation and function of anti-inflammatory Treg cells. This shifts the immune profile away from tissue inflammation, offering a unique potential for sustained, drug-free remission after stopping therapy.
Trial design: The Phase 1 trial evaluated four soquelitinib dose cohorts against placebo in 72 adult patients with moderate-to-severe atopic dermatitis who had failed at least one prior topical or systemic therapy. Notably, the study included highly refractory individuals: 35% of the total population (and 50% of the highest-dose cohort) had previously failed advanced systemic therapies, such as the biologic dupilumab (Dupixent) or Janus kinase (JAK) inhibitors.
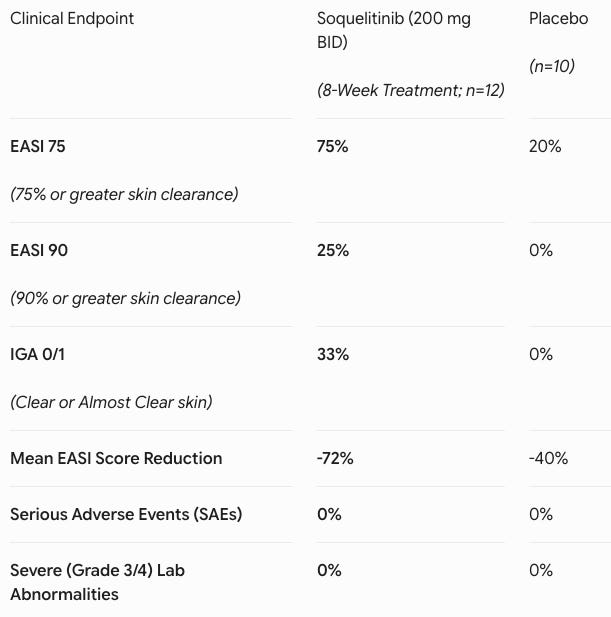
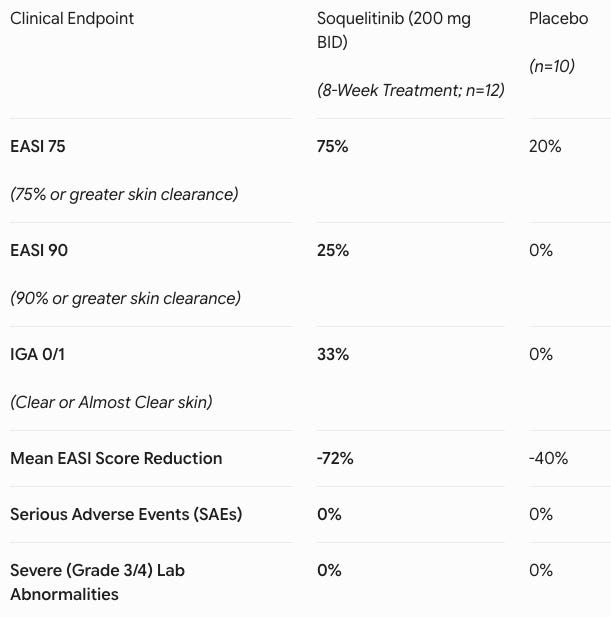
Data (press release, slide deck): The study demonstrated that extending treatment out to 8 weeks in Cohort 4 (n=12 active, n=10 evaluable placebo) significantly intensified clinical clearance (see table below). Adverse events (AEs) were low-rate and matched placebo (41.7% in both groups). Every reported AE on active drug was Grade 1 or 2 (e.g., mild nausea). There were zero serious adverse events (SAEs), zero safety-related discontinuations, and zero laboratory abnormalities.
Impact:
Oral Convenience Play: The systemic atopic dermatitis market is currently bifurcated between highly safe but injectable biologics (e.g., dupilumab, tralokinumab) and highly effective but safety-warned oral JAK inhibitors (e.g., upadacitinib, abrocitinib), which carry black-box warnings for malignancy and cardiovascular events. Soquelitinib disrupts this landscape by delivering the convenience of an oral pill paired with a clean, biologic-like safety profile. Achieving a 75% EASI 75 response in a cohort heavily populated by patients who had already failed Dupixent and JAK inhibitors highlights its potential to capture a dominant second- and third-line systemic market share. Furthermore, this success provides crucial clinical proof-of-concept for Corvus’ broader ITK-inhibitor platform across oncology and other large autoimmune indications.
Next steps: Backed by these compelling Phase 1 data, Corvus has rapidly advanced the program into a larger Phase 2 framework (SIERRA1 trial). The study targets an enrollment of approximately 200 patients with moderate-to-severe AD who are refractory to topical or systemic therapies. To refine the optimal dose, patients will be randomized across four parallel arms (~50 patients per arm): 200 mg QD, 200 mg BID, 400 mg QD, or placebo. Reflecting standard registrational trial design, the treatment timeline has been elongated to a 12-week dosing period, followed by a 90-day drug-free follow-up window to formally map and validate soquelitinib’s unique capacity for long-term, drug-free disease remission.
Descriptive data releases without numerical data
Merck - Sacituzumab Tirumotecan (TROP2 ADC) / Phase 3 (endometrial cancer): Kelun-Biotech and Merck & Co. (MSD) have announced positive topline results from the pivotal Phase 3 trial evaluating Sacituzumab Tirumotecan (sac-TMT), their investigational TROP2-directed antibody-drug conjugate (ADC), in patients with advanced endometrial cancer. The independent data monitoring committee confirmed that Sacituzumab Tirumotecan met both its primary and key secondary survival endpoints with high statistical significance (p < 0.0001). Sac-TMT demonstrated a statistically significant and clinically meaningful extension in PFS compared to TPC, substantially reducing the risk of disease progression or death. The trial successfully hit its OS endpoint, showing a clear, statistically significant survival advantage for patients treated with the ADC over standard doxorubicin or paclitaxel. Efficacy benefits were observed across all pre-specified patient subgroups, irrespective of mismatch repair status (pMMR vs. dMMR) or specific histological subtypes (endometrioid vs. serous carcinomas). The safety profile of Sacituzumab Tirumotecan was “manageable”, according to the trial sponsor, and consistent with previous Phase 2 trials. The most common treatment-related adverse events were hematologic (neutropenia, anemia, leukopenia) and gastrointestinal (nausea, diarrhea), which were effectively mitigated using standard supportive care and dose modifications. Crucially, rates of severe interstitial lung disease (ILD), a known class risk for some ADCs, remained exceptionally low. The detailed efficacy figures, including median PFS/OS months and Kaplan-Meier curves, will be presented at an upcoming major medical congress later this year (such as the ESMO or IGCS Congress). Kelun-Biotech and Merck plan to submit these data packages to global regulatory bodies, including the FDA and China’s NMPA, to secure commercial marketing authorization for this indication. Seeking to shift the asset into earlier lines of treatment, clinical trials are being initiated to evaluate Sacituzumab Tirumotecan in combination with immune checkpoint inhibitors (such as pembrolizumab) as a first-line maintenance therapy for advanced endometrial cancer.
To contact us, please send us an email at biotechreadout@gmail.com
Disclaimers
Investigational Status Disclaimer
The therapeutic candidates discussed in this newsletter are currently in clinical development and have not been approved for commercial sale by the U.S. Food and Drug Administration (FDA), the European Medicines Agency (EMA), or other global regulatory authorities. Their safety and efficacy have not been established. References to pipeline products and ongoing clinical trials involve significant risks and uncertainties. Statements regarding the potential safety, potency, or efficacy of investigational drugs reflect current hypotheses and are not a guarantee of future performance or regulatory clearance. The outcome of clinical trials is inherently unpredictable, and clinical results from earlier stages may not be predictive of results in later, larger-scale trials.
No Medical Advice Disclaimer
This newsletter is for informational and educational purposes only. The content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or another qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read in this publication.
No Patient-Provider Relationship Disclaimer
The information provided in this newsletter is for educational and analytical purposes only. Receipt of this information, or any interaction with this content, does not create a physician-patient, pharmacist-patient, or any other professional-provider relationship between you and the authors or publishers. This newsletter should not be used as a substitute for a personal consultation with a qualified healthcare professional.
Forward-Looking Statements Disclaimer
This newsletter contains “forward-looking statements” regarding future events, including clinical trial timing, regulatory milestones, and projected market performance. These statements are based on current expectations and assumptions that are subject to significant risks and uncertainties. Actual results may differ materially from those expressed or implied. We undertake no obligation to update these statements as a result of new information or future developments.
Third-Party Links & Content Disclaimer
This newsletter contains links to third-party websites, including clinical trial registries and corporate presentations. Biotech Readout does not endorse, guarantee, or assume responsibility for the accuracy or reliability of any information offered by third-party providers.
Errors and Omissions Disclaimer
While we strive for technical accuracy, the information in this newsletter is provided on an “as is” basis with no guarantees of completeness, accuracy, or timeliness. Biotech Readout assumes no liability for any errors or omissions in the content of this publication.
Non-Endorsement Disclaimer
Any reference to specific commercial products, processes, or services by trade name, trademark, or manufacturer does not constitute or imply an endorsement or recommendation by the author. All trademarks are the property of their respective owners.
No Investment Advice Disclaimer
This newsletter is for informational purposes only and does not constitute financial, investment, or legal advice. The author is not a registered investment advisor. You should consult with a professional financial advisor before making any investment decisions. The biotechnology sector is highly volatile; past performance is not indicative of future results.
Conflict of Interest Disclaimer
The author of this newsletter maintains a position of independence. At the time of publication, the author holds no direct financial interest, equity, or options in any of the companies mentioned in this report. No compensation has been received from any third party to feature or analyze specific therapeutic candidates or corporate entities.