
Weight loss maintenance strategies, 70-year hunt for metformin's mechanism, and more
Weekly Readout #7: Week of May 11, 2026

Disclaimer: This newsletter is for educational and informational purposes only and does not constitute medical, investment, or financial advice, nor does it establish a provider-patient relationship. Content may include forward-looking statements and discussions of investigational therapeutic candidates that are not FDA/EMA approved; their safety and efficacy remain unestablished and clinical outcomes are unpredictable. While we strive for accuracy, all information is provided as is without guarantees. This newsletter is independent, and the author holds no financial positions in the companies mentioned nor receives third-party compensation for this coverage. Please find a complete version of our disclaimers at the bottom of this article and on our About page.

Press ⬇️ to go down to a section and ⬆️ to go back up to the Table of Contents
Table of Contents
This week, we discuss…
…key data releases from the 33rd European Congress on Obesity (ECO):
Eli Lilly / Weight loss maintenance with Foundayo (oral GLP-1 agonist) / Phase 3b ATTAIN-MAINTAIN (obesity) ⬇️
Eli Lilly / Weight loss maintenance with low dose Zepbound (injectable GLP-1 agonist) / Phase 3b SURMOUNT-MAINTAIN (obesity) ⬇️
…two pathbreaking publications that merit a closer look (round of applause for academia this week!):
Generic / Metformin (mechanism opaque) / Nature Metabolism paper revealing its mechanism after 70 years of opacity (T2D) ⬇️
Preclinical proof of concept for rare disease with limited treatment options; Dr. David Liu Lab / ABE8e-V106W-VRQR (base editor) / Preclinical (Dravet syndrome with SCN1A-p.R613X mutation) ⬇️
…and other clinical data releases:
Potentially best-in-disease; Inhibrx / INBRX-106 (hexavalent OX40 agonist) / Phase 2 (HNSCC) ⬇️
AstraZeneca / Eneboparatide (PTH analog) / Phase 3 (hypoparathyroidism) ⬇️
Potentially best-in-class; Regenxbio / RGX-202 (AAV8-microdystrophin) / Phase 3 (DMD) ⬇️
Alkermes / Lumryz (ER sodium oxybate) / Phase 3 (idiopathic hypersomnia) ⬇️
Cabaletta / Rese-cel (CD19-CAR T) / Phase 1 (PV) ⬇️
Eli Lilly / Weight loss maintenance with Foundayo (oral GLP-1 agonist) / Phase 3b (obesity)
The ATTAIN-MAINTAIN Phase 3b clinical trial evaluated Foundayo (orforglipron), a first-in-class, oral, non-peptide GLP-1 receptor agonist, as a maintenance therapy for individuals who achieved initial weight loss using injectable GLP-1 medications like Wegovy (semaglutide) or Zepbound (tirzepatide). The data was officially presented today (May 12, 2026) at the 33rd European Congress on Obesity (ECO) held in Istanbul and the full study was simultaneously published in Nature Medicine.
Indication: We covered the obesity in a two-part Frontiers in Medicine series (Part 1, Part 2).
Mechanism: Foundayo is a small-molecule GLP-1 receptor agonist. Unlike existing peptide-based oral options (e.g., Rybelsus, oral Wegovy), it does not require complex absorption enhancers or strict fasting/water restrictions. It stimulates insulin secretion, suppresses glucagon, slows gastric emptying, and reduces appetite by acting on brain centers that regulate satiety.
Trial design: The ATTAIN-MAINTAIN trial utilized a “switch” design to test the sustainability of weight loss when transitioning from injectable to oral therapy. It included adults with obesity or overweight (without Type 2 Diabetes) who had completed a 52-week weight loss induction phase on either injectable Wegovy (2.4 mg) or Zepbound (15 mg). After the induction phase, participants were re-randomized to receive once-daily Foundayo (orforglipron) or a placebo for an additional 52 weeks of maintenance. The primary endpoint was the percentage change in body weight from the start of the maintenance period (Week 52) to the end of the trial (Week 104).
Data (press release, Nature Medicine paper): The trial demonstrated that Foundayo effectively maintained the weight loss achieved on injectables (see table below). Those who switched to Foundayo regained an average of only 0.9 kg over the year, compared to significant weight regain typically seen in placebo groups after stopping injectables. The most common adverse events were gastrointestinal (nausea, vomiting).
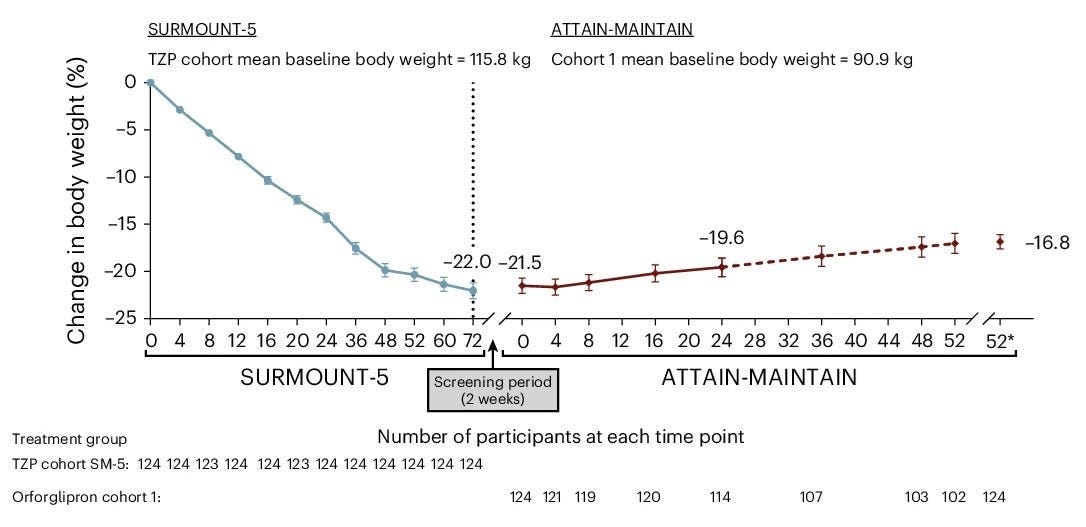
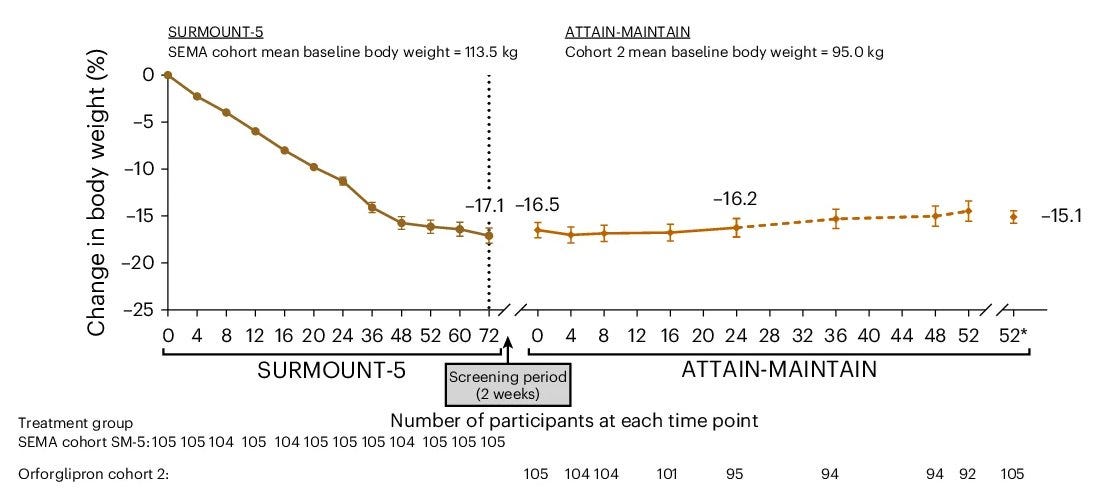
Impact:
Foundayo for Weight Loss Maintenance: It provides a clinically viable exit strategy for patients who want to transition off weekly injections but fear the rebound weight gain associated with treatment cessation.
Oral Convenience: Foundayo removes the needle phobia and the fasting requirement associated with other GLP-1 therapies. It can be taken at any time of day, with or without food.
Next steps: Following its FDA approval on April 1, 2026, the manufacturer is pursuing regulatory filings Foundayo in the UK (MHRA) and EU (EMA). The trial sponsor telegraphed than future studies will focus on the 3- to 5-year durability of maintenance to see if patients can remain on a low-dose oral maintenance pill indefinitely. Data from the separate ATTAIN-CVOT trial will be monitored to confirm if Foundayo provides the same heart-protective benefits as injectable Wegovy.
Eli Lilly / Weight loss maintenance with low dose Zepbound (injectable GLP-1 agonist) / Phase 3b (obesity)
The SURMOUNT-MAINTAIN Phase 3b trial results, officially presented today (May 12, 2026) at the European Congress on Obesity (ECO) and published in The Lancet, provide critical evidence for the “maintenance phase” of obesity treatment. This study addresses a key clinical question: Can patients maintain their weight loss on a lower “maintenance dose” of Zepbound (tirzepatide)?
Indication: We covered the obesity in a two-part Frontiers in Medicine series (Part 1, Part 2).
Mechanism: Zepbound (tirzepatide) is a dual GIP and GLP-1 receptor agonist. GIP potentiates insulin secretion and acts on the brain to improve lipid metabolism and potentially reduce the nausea often associated with GLP-1 alone. GLP-1 delays gastric emptying and increases satiety. The combination leads to superior weight loss compared to single GLP-1 agonists by targeting multiple metabolic pathways and appetite centers in the hypothalamus.
Trial design: The SURMOUNT-MAINTAIN Phase 3b trial focused on dose-reduction as a long-term strategy for weight management. It included 441 participants with obesity or overweight (without diabetes) who received 60 weeks of open-label Zepbound, titrated to their Maximum Tolerated Dose (MTD), either 10 mg or 15 mg. Those who achieved a weight plateau (losing ≥5% of body weight) were re-randomized for 52 additional weeks into three arms:
Data (press release, Lancet paper): The trial demonstrated a clear dose-response relationship for maintaining weight loss (see graph below). Discontinuation due to adverse events during the maintenance phase was extremely low: 0% for MTD and 0.7% for the 5 mg dose. Nausea and vomiting were significantly lower in the 5 mg group compared to those staying on the higher MTD.
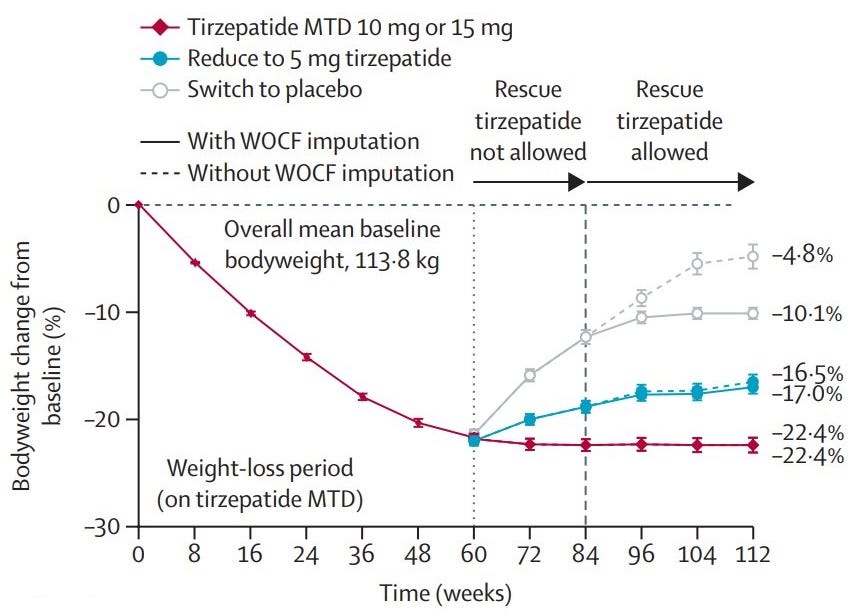
Impact:
“Step-Down” Strategy: The “placebo rebound” (regaining over half of the lost weight) confirms that obesity is a chronic condition requiring long-term pharmacotherapy. For the first time, clinicians have high-quality data showing that 5 mg is a viable “maintenance pill” (or injection). While it may result in a small amount of regain, it drastically improves tolerability and remains far superior to stopping treatment entirely.
Economic Benefit: Using a lower dose for maintenance could potentially reduce the long-term cost of treatment and improve supply chain availability for the higher-dose pens.
Next steps: Eli Lilly is expected to use this data to update the Zepbound prescribing information to include specific maintenance dosing guidelines. Next-stage trials are monitoring Morbidity and Mortality in Obesity (SURMOUNT-MMO) to see if the 5 mg dose provides the same long-term cardiovascular and renal protection as the higher doses.
Generic / Metformin (mechanism opaque) / Nature Metabolism paper revealing its mechanism (T2D)
Indication: Type 2 diabetes (T2D) is characterized by a dual failure of glucose regulation: peripheral insulin resistance, where cells (muscle, liver, and fat) stop responding effectively to insulin, and a compensatory beta-cell dysfunction where the pancreas can no longer produce enough insulin to overcome that resistance. This results in chronic hyperglycemia, which can damage blood vessels and organs over time. Metformin remains the first-line gold standard of care, despite our limited understanding of its mechanism of action, because it addresses this pathology without causing weight gain or hypoglycemia.
Mechanism: The mechanism of metformin has been considered clinically opaque for approximately 70 years, dating back to its first use for diabetes in the mid-1950s. While metformin is the most widely prescribed drug for Type 2 Diabetes (T2D), it was discovered before the era of target-based drug design. For decades, the medical community accepted that it worked, even as they debated exactly how. Here is a brief timeline describing the opacity:
1922–1957 (The Origins): Metformin was first synthesized in 1922 but sat in obscurity for decades. It was finally introduced as a diabetes treatment in 1957 by Jean Sterne.
1994–2000 (The Mitochondrial Theory): Researchers began to suspect the liver was the primary target. In 2000, two independent groups proposed that it works by inhibiting Complex I of the mitochondrial electron transport chain.
2001–2020 (The AMPK Debate): For nearly 20 years, the leading theory was that it worked through the AMPK pathway. However, this was later challenged by “loss-of-function” studies showing the drug still worked even without AMPK, throwing the mechanism back into uncertainty.
2025 (The “Brain Pathway” Breakthrough): In July 2025, researchers have identified a “hidden” neural mechanism. Studies published in Science Advances revealed that metformin suppresses a protein called Rap1 in the hypothalamus, activating specific neurons to lower blood sugar.
Summary of the old theory for how metformin worked: For more than three decades, the medical community has operated under a comfortable consensus: Metformin, the world’s most widely prescribed diabetes medication, works by targeting the liver. The narrative was simple and elegant. The drug enters the body, travels to the liver, and shuts down the production of excess glucose (gluconeogenesis). However, a persistent molecular mystery remained beneath the surface. Clinical data increasingly showed that standard doses of metformin rarely reach the liver in concentrations high enough to actually inhibit its molecular targets. Even more puzzling, recent studies noted that metformin fails to reduce glucose production in patients with well-controlled or recent-onset type II diabetes—yet the drug remains remarkably effective. If the liver isn’t the primary battlefield, where is the war against high blood sugar actually being won?
New Data (Nature Metabolism paper): The key finding in the new research involves how metformin tricks the body into disposing of sugar. Using FDG-PET imaging, the researchers observed that metformin causes the small and large intestines to light up with activity. This happens because metformin inhibits mitochondrial complex I specifically within the intestinal epithelium (the gut lining). When these mitochondria are suppressed, the gut cells face an immediate energy crisis. To compensate, they stop relying on efficient oxygen-based energy production and switch to a high-speed, glucose-burning mode called glycolysis. Essentially, the drug turns the gut into a “glucose sink.” Instead of sugar passing through the intestinal wall and into the bloodstream, the gut cells pull massive amounts of glucose out of circulation to fuel their own survival.
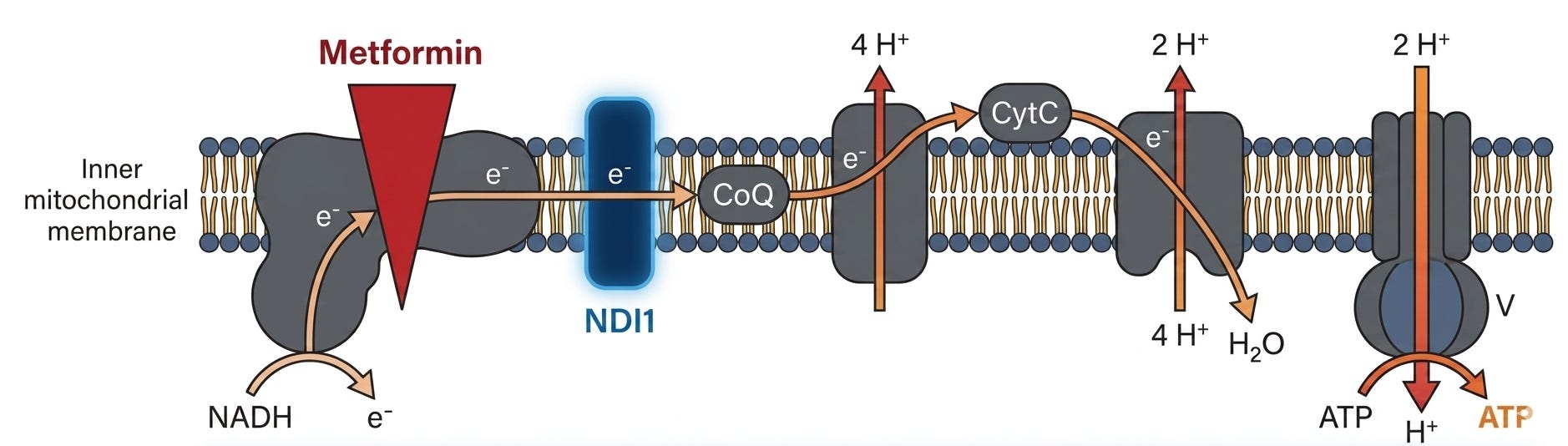
Impact:
Potential for “Better Metformin”: The study suggests that the future of biguanide therapy may lie in gut-restricted formulations that keep the drug’s concentration high in the intestines while minimizing systemic absorption. By combining gut-targeted drugs with P-glycoprotein inhibitors (like encequidar), researchers achieved a “far more dramatic” glucose-lowering effect with a similar drug (berberine). This approach could be used to develop new, more powerful metformin-like therapies that act exclusively in the “glucose sink” of the gut without causing toxicity in other organs.
Dr. David Liu Lab / ABE8e-V106W-VRQR (base editor) / Preclinical (Dravet syndrome with SCN1A-p.R613X mutation)
In a study published in Science Translational Medicine on May 13, 2026, researchers from the David Liu Lab at the Broad Institute of MIT and Harvard, in collaboration with The Jackson Laboratory, demonstrated a potentially transformative approach to treating Dravet syndrome in genetic mouse models and human cells lines that mimic the disease.
Indication: Dravet Syndrome (DS) is a catastrophic neurodevelopmental channelopathy characterized by treatment-resistant epilepsy, temperature-sensitive seizures, and a high incidence of sudden unexpected death in epilepsy (SUDEP). Approximately 80% to 90% of cases arise from de novo loss-of-function variants in SCN1A, the gene encoding the voltage-gated sodium channel α subunit Nav1.1. While current pharmacological management addresses symptomatic seizure burden, it fails to remediate the underlying genetic haploinsufficiency. In humans, DS symptoms typically manifest between 3 and 6 months of age, coinciding with the developmental surge in SCN1A expression. This temporal window defines a critical opportunity for disease prevention. The molecular pathology of the SCN1A-p.R613X variant centers on the impairment of parvalbumin-expressing (PV) inhibitory interneurons, where Nav1.1 is preferentially expressed. The introduction of a premature stop codon triggers nonsense-mediated decay (NMD) of the mutant transcript, leading to a profound reduction in Nav1.1 protein levels. This deficiency causes PV-interneuron hypoexcitability, disrupting the cortical inhibitory-excitatory balance and precipitating the DS phenotype.
Mechanism: The ABE8e-V106W-VRQR is a precision genome editing tool designed to correct genetic mutations by converting A·T base pairs to G·C at specific locations without requiring double-stranded DNA breaks or donor DNA templates. Its mechanism is defined by the synergy of its three primary components: a highly active deaminase (ABE8e), a precision-enhancing mutation (V106W), and a flexible DNA-binding Cas9 domain (VRQR). To treat Dravet syndrome, the ABE8e-V106W-VRQR was used to target a specific Arg-to-STOP mutation (CGA > TGA). The editor binds to the target site using the VRQR domain guided by an sgRNA, and the V106W-modified ABE8e deaminase converted the stop codon (TGA) back into the functional arginine codon (CGA). This direct correction is designed to restore the production of functional Nav1.1 sodium channels, addressing the root cause of the disease. The system achieved 97% mRNA editing efficiency in the mouse neocortex when administered via one intracerebroventricular (ICV) injection, successfully targeting Parvalbumin-positive inhibitory neurons (PVINs).
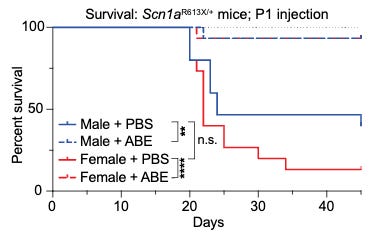
Data (Science Translational Medicine paper):
Outcomes of treated Dravet mice: In the 42.5°C heat lamp test (simulating a high fever), 100% of ABE-treated mice remained seizure-free, whereas vehicle-treated controls suffered seizures at an average of 40°C. 24-hour video monitoring showed that treatment entirely eliminated spontaneous seizures in treated animals, while 50–60% of untreated mice experienced frequent events. Treated Dravet mice achieved a 90% survival rate at Day 45 versus 27% survival in untreated mice.
Whole-cell patch-clamp recordings of Dravet mouse neurons: The team demonstrated that ABE treatment successfully rebalanced the brain’s circuitry. By correcting the sodium current and firing kinetics of these interneurons, the treatment successfully reversed the “electrical storms” of the Dravet brain, restoring the inhibitory brakes needed to prevent paroxysmal activity.
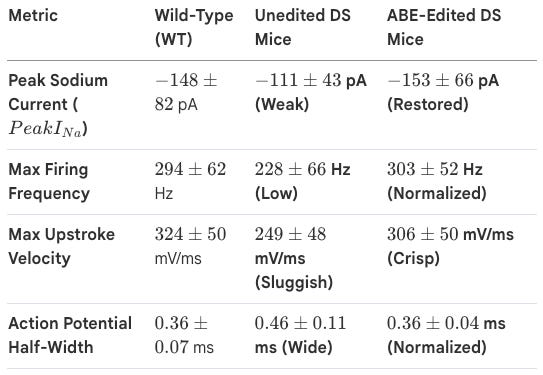
Impact: Note that this approach is neither approved for use in human patients, nor is there any indication that it will enter clinical trials (as of May 2026). Before this can be applied to human patients, several areas require further optimization.
Optimization for Other SCN1A Variants: Because base editing is a precision tool, different strategies must be optimized for each specific genetic variant found in DS patients. . However, over 1,400 pathogenic SCN1A variants are currently identified that could potentially be targeted by precision genome editing like base or prime editing.
Advanced Delivery Methods: While this study used AAV9, future human applications may benefit from more transient delivery modalities, such as lipid nanoparticles (LNPs) or engineered virus-like particles (eVLPs), which could further reduce the risk of long-term off-target editing. Optimization for in human pharmacokinetics and safety would require Phase 1 clinical trials at least.
Early Genetic Screening: The researchers emphasize that early genetic screening and the use of pathogenicity-scoring tools (like AlphaMissense) will be essential to identify candidates for intervention before severe symptoms manifest.
Inhibrx / INBRX-106 (hexavalent OX40 agonist) / Phase 2 (HNSCC)
The Phase 2/3 HexAgon-HN trial results represent a potential breakthrough for INBRX-106, a first-in-class hexavalent OX40 agonist, particularly in a landscape where previous bivalent OX40 candidates have historically failed to show clinical efficacy.
Indication: Head and Neck Squamous Cell Carcinoma (HNSCC) primarily arises from the mucosal epithelium of the oral cavity, pharynx, and larynx, driven by genomic instability from chronic carcinogen exposure (tobacco and alcohol) or high-risk Human Papillomavirus (HPV) infection. Pathophysiologically, these insults lead to the loss of tumor suppressors like p53 and p16, alongside the upregulation of growth factor receptors such as EGFR, creating an immunosuppressive microenvironment that facilitates local invasion and lymphatic spread. The standard of care is risk-stratified: early-stage disease is typically managed with single-modality surgery or radiation, while locally advanced cases require a multidisciplinary approach involving surgery followed by adjuvant chemoradiotherapy or definitive cisplatin-based radiation. For recurrent or metastatic disease, the paradigm has shifted toward immune checkpoint inhibitors (e.g., pembrolizumab) with or without platinum-based chemotherapy, depending on PD-L1 expression levels.
Mechanism: Unlike traditional bivalent antibodies that typically require Fc-receptor cross-linking to activate their targets, INBRX-106 is engineered as a hexavalent molecule. It induces higher-order clustering of the OX40 (CD134) receptor, which is critical for potent downstream signaling. OX40 is a tumor necrosis factor receptor (TNFR) family member expressed on activated CD4+ and CD8+ T cells. Agonism leads to enhanced T-cell survival, proliferation, and effector function. Combining INBRX-106 with Keytruda (pembrolizumab) addresses checkpoint resistance by providing a positive costimulatory “gas pedal” (OX40) while simultaneously removing the “brakes” (PD-1).
Trial design: The data stems from the randomized, open-label Phase 2/3 HexAgon-HN study evaluating INBRX-106 in combination with Keytruda versus Keytruda alone in first-line (1L) patients with recurrent or metastatic (R/M) Head and Neck Squamous Cell Carcinoma (HNSCC). Patients were required to have a PD-L1 Combined Positive Score (CPS) ≥20.
Data (press release): The reported readout shows a significant doubling of the objective response rate (ORR) compared to the current standard of care (44% for INBRX-106 + Keytruda versus 21.4% for Keytruda monotherapy). Early signals suggest the combination is generally well-tolerated, with most adverse events being Grade 1 or 2, avoiding the severe toxicity often seen with high-dose cytokine or early-generation co-stimulatory agents.
Impact:
Overcoming “Cold” Tumors: HNSCC is highly immunosuppressive; the 44% ORR suggests that hexavalent OX40 agonism can successfully prime the immune system in patients who would otherwise have a limited response to PD-1 inhibitors alone.
Potentially Best-in-Indication: This data places INBRX-106 at the forefront of the “next generation” of HNSCC therapies, potentially challenging the current 1L standard of care for high PD-L1 expressing patients.
Next steps: Following the success of the Phase 2 portion, the trial is designed to transition into a Phase 3 registrational study to confirm these ORR results and evaluate long-term endpoints like Progression-Free Survival (PFS) and Overall Survival (OS). If the Phase 3 data remains consistent, Inhibrx (under Sanofi, following their acquisition) will likely seek FDA approval for INBRX-106 as a 1L combination therapy for HNSCC.
AstraZeneca / Eneboparatide (PTH analog) / Phase 3 (hypoparathyroidism)
AstraZeneca recently reported positive topline results for the Phase 3 CALYPSO trial evaluating eneboparatide (AZP-3601) in adults with chronic hypoparathyroidism. The data, presented at the European Congress of Endocrinology (ECE) 2026 in Prague, marks a significant step forward for AstraZeneca’s rare disease division, Alexion.
Indication: The pathophysiology of hypoparathyroidism is defined by the absolute or relative deficiency of parathyroid hormone (PTH), which disrupts systemic mineral homeostasis. Under normal conditions, PTH stimulates bone resorption, distal renal calcium reabsorption, and the conversion of vitamin D to its active form (1,25(OH)2D) to increase intestinal calcium absorption; its absence lead to the biochemical triad of hypocalcemia, hyperphosphatemia, and hypercalciuria. While most cases are acquired (post-surgical damage during neck surgery), others are autoimmune or genetic (e.g., CaSR or GATA3 mutations). The standard of care has historically relied on “conventional therapy” with high-dose oral calcium and active vitamin D (calcitriol), but this approach often fails to address long-term complications such as nephrocalcinosis and chronic kidney disease. Consequently, recent 2025-2026 clinical guidelines have shifted toward PTH replacement therapy, specifically with the FDA-approved Yorvipath (palopegteriparatide), which provides a more physiologic replacement that stabilizes serum calcium while reducing the risk of renal damage and improving patient quality of life.
Mechanism: Eneboparatide is a novel, synthetic peptide designed as a selective PTH1 receptor agonist. It binds with high affinity to the R0 conformation of the PTH1 receptor. This specific binding triggers a sustained cyclic AMP (cAMP) signal, which more closely mimics physiological PTH activity compared to earlier analogs.conformation of the PTH1 receptor. This specific binding triggers a sustained cyclic AMP (cAMP) signal, which more closely mimics physiological PTH activity compared to earlier analogs.onformation of the PTH1 receptor. This specific binding triggers a sustained cyclic AMP (cAMP) signal, which more closely mimics physiological PTH activity compared to earlier analogs. It is engineered to maintain serum calcium (sCa) within the normal range while simultaneously increasing renal calcium reabsorption (reducing urinary calcium) and maintaining a balanced resumption of bone turnover without causing excessive bone resorption.
Trial design: CALYPSO is the largest global Phase 3 trial conducted in 202 adults with chronic hypoparathyroidism receiving standard of care (oral calcium and active vitamin D). Patients were randomized 2:1 (eneboparatide vs. placebo) for a 24-week double-blind treatment period. Patients in the treatment arm were dosed with once-daily subcutaneous injections of eneboparatide starting at 20 µg, with titration up to 100 µg. The trial’s composite endpoint was achievement of normal albumin-adjusted serum calcium (8.3–10.6 mg/dL) AND independence from conventional therapy (elimination of active vitamin D and ≤600 mg/day of oral calcium).
Data (press release): The trial met its primary and all key secondary endpoints with high statistical significance (p = 0.0001).
Primary Endpoint (Normalization + Independence): 31.1% of eneboparatide patients met the composite goal at Week 24 vs. 5.9% in the placebo group.
Secondary Endpoint (Hypercalciuria): Among patients with high urinary calcium at baseline, 56.6% normalized their levels on eneboparatide vs. 20% on placebo.
Bone Health: Biomarkers (P1NP and CTX) remained within normal ranges, and there was no clinically significant decline in bone mineral density (BMD) through 52 weeks.
Safety: Generally well-tolerated. While immunogenicity (anti-drug antibodies) was observed in a majority of patients, the company noted that calcium control could still be maintained through dose titration.
Impact:
Renal Protection: By specifically reducing urinary calcium excretion (hypercalciuria), eneboparatide addresses one of the most significant long-term risks of hypoparathyroidism, chronic kidney disease and nephrocalcinosis.
Quality of Life: Significant improvements were reported in disease-specific physical symptoms and physical functioning scores (SF-36).
Physiologic Balance: Its design aims to avoid the bone-stripping effect sometimes seen with continuous high-dose PTH exposure, making it a potentially safer long-term option for skeletal integrity.
Next steps: Detailed results from the 52-week open-label extension (which showed sustained benefits) were presented alongside the primary data. AstraZeneca plans to move forward with global regulatory submissions. Eneboparatide will likely compete with Ascendis Pharma’s Yorvipath (palopegteriparatide), which was approved a couple of years ago on August 12, 2024.
Regenxbio / RGX-202 (AAV8-microdystrophin) / Phase 3 (DMD)
On May 14, 2026, REGENXBIO announced pivotal topline results for RGX-202 in Duchenne Muscular Dystrophy (DMD). While the trial hit its primary biomarker endpoint with high statistical significance, the market’s reaction has been tempered by a reported case of myocarditis and broader skepticism regarding single-arm gene therapy approvals.
Indication: Duchenne muscular dystrophy (DMD) is an X-linked recessive genetic disorder characterized by the absence or functional deficiency of dystrophin, a critical protein that anchors the cytoskeleton of muscle fibers to the surrounding extracellular matrix. Pathophysiologically, the lack of dystrophin renders the sarcolemma fragile, leading to membrane rupture during muscle contraction, chronic calcium influx, and a cascade of inflammation and oxidative stress that results in the progressive replacement of muscle tissue with fibrosis and adipose tissue. The standard of care focuses on multidisciplinary management, primarily utilizing long-term corticosteroid therapy (e.g., prednisone or deflazacort) to delay the loss of ambulation and preserve respiratory and cardiac function. In recent years, the paradigm has expanded to include mutation-specific exon-skipping therapies and the first FDA-approved gene therapy, Elevydis (delandistrogene moxeparvovec-rokl), which aims to introduce a functional micro-dystrophin transgene to address the underlying genetic cause.
Mechanism: RGX-202 uses an AAV8 vector to deliver a proprietary microdystrophin transgene. It is currently the only clinical-stage gene therapy that includes the C-Terminal (CT) domain. The CT domain is designed to recruit key proteins to the muscle cell membrane, potentially providing superior structural integrity and protection against contraction-induced damage compared to truncated versions like Sarepta’s Elevydis.
Trial design: The data comes from the pivotal Phase 3 portion of a seamless Phase 1/2/3 multicenter, open-label (single-arm) AFFINITY DUCHENNE trial. It included 31 ambulatory boys (aged 1–12 years) received the pivotal dose of 2 × 1014 GC/kg. The primary endpoint was microdystrophin expression levels at 12 weeks. Secondary endpoints included functional changes measured by the North Star Ambulatory Assessment (NSAA) at 12 months.
Data (press release, slide deck): Approximately 93% of patients (28/30) achieved microdystrophin expression >10% of normal at 12 weeks (p < 0.0001), meeting the primary endpoint. Mean expression was a robust 71.1%. In an interim cut of 9 patients at 12 months, there was a statistically significant correlation (p = 0.0002) between microdystrophin levels and functional improvement (NSAA). Regenxbio claims this correlation validates the biomarker as a surrogate for clinical benefit. One case of subacute myocarditis occurred in an 8-year-old participant. While the company noted the case resolved without changes to ejection fraction or permanent fibrosis, myocarditis is a known “class risk” for AAV gene therapies that often triggers intense regulatory scrutiny. Regenxbio reported that only one patient out of 31 (~3.2%) in the pivotal cohort developed a severe liver injury. The company highlighted this as a significant edge over Sarepta’s Elevydis, which has reported liver injury rates closer to 40%. Data through the two-year mark for earlier participants shows that mean gamma-glutamyl transferase (GGT) and total bilirubin levels, sensitive markers for liver inflammation, did not exceed the upper limit of normal.
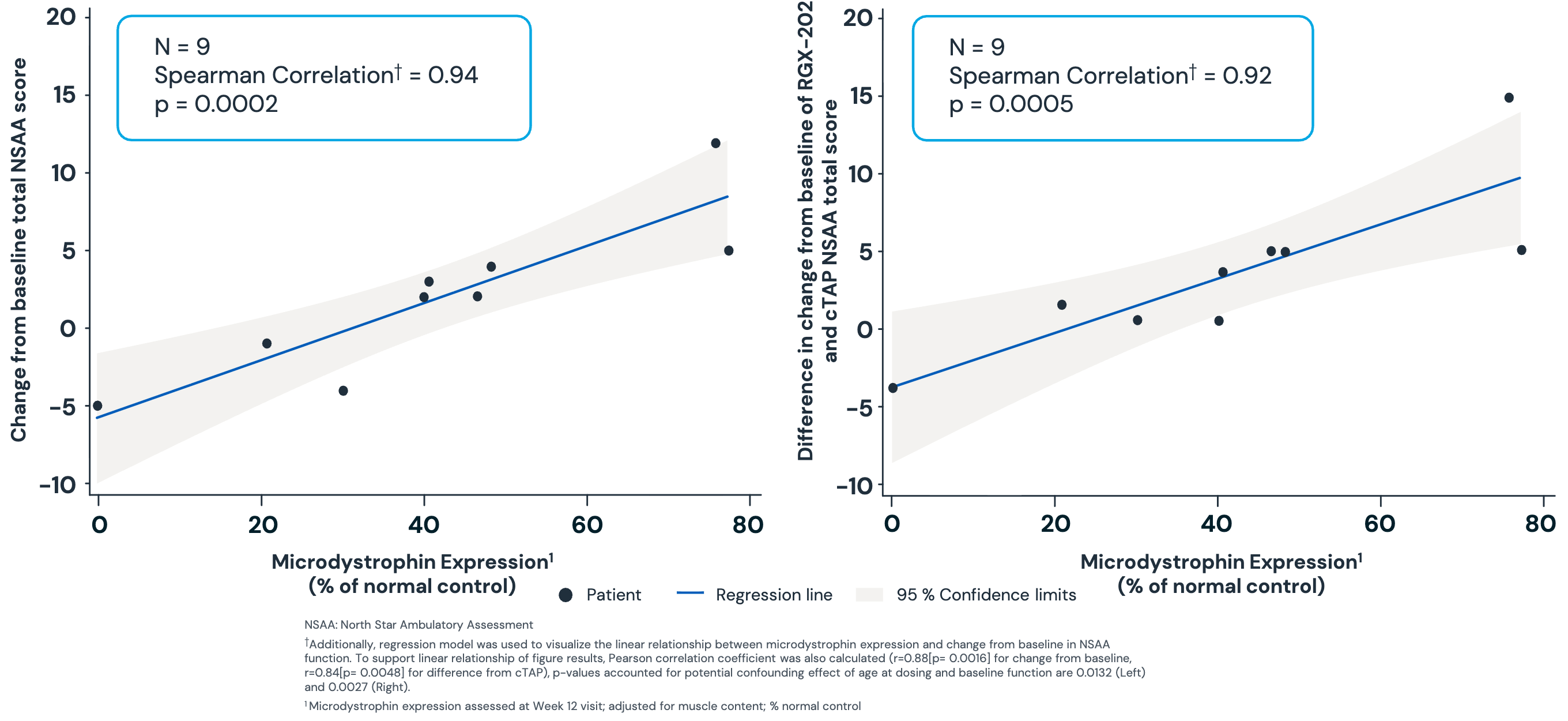
Mixed Impact:
Best-in-Class Potential: The high expression levels and the presence of the CT domain suggest RGX-202 could be more potent than first-generation therapies.
UniQure Precedent: Single-arm data failed to immediately convince the FDA of a clear functional “win” over natural history controls in the case of UniQure gene therapy for Huntington’s disease. Critics argue that without a placebo arm, it is difficult to prove the NSAA gains aren’t due to the intensive steroid regimens or patient selection.
Next steps: Regenxbio expects to complete dosing for the full 60-patient pivotal and confirmatory cohorts by mid-2026. The company plans to request a pre-BLA meeting with the FDA in mid-2026, aiming for a Biologics License Application (BLA) submission for accelerated approval in 2027.
Descriptive data releases without numerical data
Alkermes / Lumryz (ER sodium oxybate) / Phase 3 (idiopathic hypersomnia): Idiopathic hypersomnia (IH) is a chronic neurological disorder characterized by debilitating excessive daytime sleepiness (EDS), prolonged nocturnal sleep, and severe “sleep inertia” (extreme difficulty awakening). Its pathophysiology remains largely unknown and is a diagnosis of exclusion. Alkermes recently announced positive topline results for the Phase 3 REVITALYZ study of Lumryz (extended-release sodium oxybate) in adults with Idiopathic Hypersomnia (IH). This readout is a significant milestone following Alkermes’ acquisition of Avadel Pharmaceuticals earlier this year. The trial met its primary and all key secondary endpoints with high statistical significance (p < 0.0001). Patients switched to placebo experienced a statistically significant worsening of excessive daytime sleepiness as measured by the Epworth Sleepiness Scale (ESS) compared to those who stayed on Lumryz. The safety profile was consistent with previous Lumryz trials in narcolepsy. Common adverse events (≥10%) included nausea, headache, anxiety, dizziness, and vomiting; no new safety signals were identified. Alkermes plans to present the detailed, granular data from the REVITALYZ study at a medical meeting later this year and submit a supplemental New Drug Application (sNDA) to the FDA by the end of 2026.
Cabaletta / Rese-cel (CD19-CAR T) / Phase 1 (PV): Pemphigus vulgaris (PV) is a rare, life-threatening autoimmune blistering disease where the pathophysiology centers on the production of IgG autoantibodies (primarily against desmoglein 3 and/or desmoglein 1). These proteins are critical components of desmosomes, which provide the “glue” that holds keratinocytes together; the autoantibody binding triggers a process called acantholysis, leading to the separation of epidermal layers and the formation of fragile, flaccid bullae on the skin and mucous membranes. The standard of care in 2026 focuses on rapid disease control and long-term remission using a combination of systemic corticosteroids (prednisone) and the anti-CD20 monoclonal antibody rituximab, which is now firmly established as a first-line treatment for moderate-to-severe cases. Cabaletta Bio recently shared promising Phase 1 data for rese-cel (resecabtagene autoleucel) in Pemphigus Vulgaris (PV), specifically highlighting the success of their “preconditioning-free” (PC-free) approach at the ASGCT 2026 Annual Meeting (May 14, 2026). The current data focused on the first four refractory patients (mucosal or mucocutaneous PV) treated at the lowest dose (1.0 × 106 cells/kg) without preconditioning: 2 out of 4 patients (50%) maintained a “compelling drug-free response” through 6 months of follow-up after stopping all immunomodulators; 3 of the 4 patients achieved complete peripheral B cell depletion, with CAR T expansion kinetics similar to those seen in patients with preconditioning. Rese-cel was generally well-tolerated. No Dose Limiting Toxicities (DLTs) or ICANS (neurotoxicity) were reported; one patient experienced Grade 1 Cytokine Release Syndrome (transient fever). Complete Phase 1/2 data across the RESET program, including further details on RESET-PV, will be presented at the EULAR 2026 Congress (June 3-6, 2026) in London.
AstraZeneca / Imfinzi (anti-PDL1 mAb) / Phase 3 (MIBC): Muscle-Invasive Bladder Cancer (MIBC) occurs when malignant cells, primarily urothelial carcinomas, penetrate the basement membrane and invade the detrusor muscle layer (Stage T2–T4a) of the bladder wall. On May 14, 2026, AstraZeneca announced positive high-level results from the Phase 3 VOLGA trial. This marks the third successful Phase 3 readout for Imfinzi in bladder cancer (following NIAGARA and POTOMAC), solidifying its position in the curative-intent setting for patients who cannot receive standard platinum-based therapies. The interim analysis showed that the combination of Imfinzi + Padcev outperformed the current standard of care, having demonstrated statistically significant and clinically meaningful improvements in both event-free survival (EFS) and overall survival (OS) compared to the control group. The safety profile was consistent with the known profiles of each drug; no new safety signals were identified in the combination. Detailed data, including hazard ratios and secondary endpoints like Pathologic Complete Response (pCR), will be presented at an upcoming medical meeting (likely ESMO or ASCO GU).
To contact us, please send us an email at biotechreadout@gmail.com
Disclaimers
Investigational Status Disclaimer
The therapeutic candidates discussed in this newsletter are currently in clinical development and have not been approved for commercial sale by the U.S. Food and Drug Administration (FDA), the European Medicines Agency (EMA), or other global regulatory authorities. Their safety and efficacy have not been established. References to pipeline products and ongoing clinical trials involve significant risks and uncertainties. Statements regarding the potential safety, potency, or efficacy of investigational drugs reflect current hypotheses and are not a guarantee of future performance or regulatory clearance. The outcome of clinical trials is inherently unpredictable, and clinical results from earlier stages may not be predictive of results in later, larger-scale trials.
No Medical Advice Disclaimer
This newsletter is for informational and educational purposes only. The content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or another qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read in this publication.
No Patient-Provider Relationship Disclaimer
The information provided in this newsletter is for educational and analytical purposes only. Receipt of this information, or any interaction with this content, does not create a physician-patient, pharmacist-patient, or any other professional-provider relationship between you and the authors or publishers. This newsletter should not be used as a substitute for a personal consultation with a qualified healthcare professional.
Forward-Looking Statements Disclaimer
This newsletter contains “forward-looking statements” regarding future events, including clinical trial timing, regulatory milestones, and projected market performance. These statements are based on current expectations and assumptions that are subject to significant risks and uncertainties. Actual results may differ materially from those expressed or implied. We undertake no obligation to update these statements as a result of new information or future developments.
Third-Party Links & Content Disclaimer
This newsletter contains links to third-party websites, including clinical trial registries and corporate presentations. Biotech Readout does not endorse, guarantee, or assume responsibility for the accuracy or reliability of any information offered by third-party providers.
Errors and Omissions Disclaimer
While we strive for technical accuracy, the information in this newsletter is provided on an “as is” basis with no guarantees of completeness, accuracy, or timeliness. Biotech Readout assumes no liability for any errors or omissions in the content of this publication.
Non-Endorsement Disclaimer
Any reference to specific commercial products, processes, or services by trade name, trademark, or manufacturer does not constitute or imply an endorsement or recommendation by the author. All trademarks are the property of their respective owners.
No Investment Advice Disclaimer
This newsletter is for informational purposes only and does not constitute financial, investment, or legal advice. The author is not a registered investment advisor. You should consult with a professional financial advisor before making any investment decisions. The biotechnology sector is highly volatile; past performance is not indicative of future results.
Conflict of Interest Disclaimer
The author of this newsletter maintains a position of independence. At the time of publication, the author holds no direct financial interest, equity, or options in any of the companies mentioned in this report. No compensation has been received from any third party to feature or analyze specific therapeutic candidates or corporate entities.