
First successful Phase 3 for an in vivo CRISPR gene therapy, and more
Weekly Readout #5: Week of April 27, 2026

Disclaimer: This newsletter is for educational and informational purposes only and does not constitute medical, investment, or financial advice, nor does it establish a provider-patient relationship. Content may include forward-looking statements and discussions of investigational therapeutic candidates that are not FDA/EMA approved; their safety and efficacy remain unestablished and clinical outcomes are unpredictable. While we strive for accuracy, all information is provided as is without guarantees. This newsletter is independent, and the author holds no financial positions in the companies mentioned nor receives third-party compensation for this coverage. Please find a complete version of our disclaimers at the bottom of this article and on our About page.

Press ⬇️ to go down to a section and ⬆️ to go back up to the Table of Contents
Table of Contents
This week, we discuss:
Intellia / Lonvo-z (KLKB1 KO) / Phase 3 (HAE) - the first-ever successful Phase 3 readout for an in vivo CRISPR gene-editing therapy ⬇️
Oruka / ORKA-001 (IL-23p19 mAb HLE) / Phase 2a (plaque psoriasis) - potential best-in-indication therapy (once yearly injectable versus current standard of 4–6 injections per year) ⬇️
Veradermics / VDPHL01 (ER oral minoxidil) / Phase 2/3 (androgenetic alopecia) - if approved, it would be the first FDA-sanctioned oral non-hormonal treatment for hair loss in nearly 30 years ⬇️
Mirum / Brelovitug (anti-HBsAg mAb) / Phase 2b (Hepatitis D) ⬇️
Incyte / Povorcitinib (JAK1 inhibitor) / Phase 3 (nonsegmental vitiligo) ⬇️
Intellia / Lonvo-z (KLKB1 KO) / Phase 3 (HAE)
On April 27, 2026, Intellia Therapeutics announced positive topline results for the Phase 3 HAELO trial of lonvoguran ziclumeran (lonvo-z), formerly NTLA-2002. This marks the first-ever successful Phase 3 readout for an in vivo CRISPR gene-editing therapy.
Indication: I covered the causal mechanism and treatment landscape of hereditary angioedema (HAE) in my piece on the Chiesi-KalVista acquisition earlier this week. Standard of care is gradually transitioning from the injectable C1-INH replacement (Cinryze) and anti-kallikrein mAbs (Takhzyro) to oral plasma kallikrein inhibitors (Orladeyo, Ekterly).
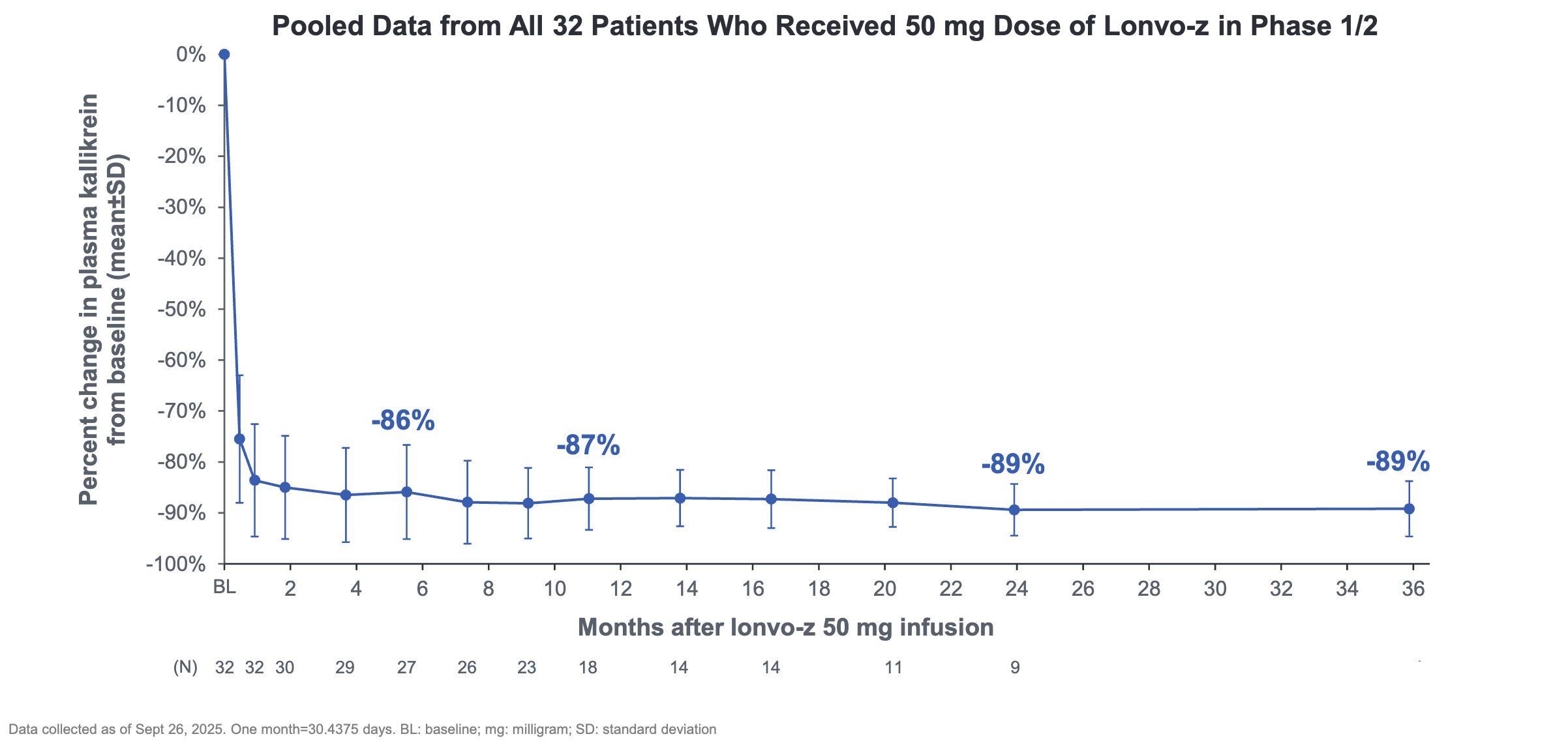
Mechanism: Lonvo-z is an in vivo CRISPR/Cas9 therapy delivered via lipid nanoparticles (LNPs) that target the liver. It permanently inactivates the KLKB1 gene in hepatocytes, preventing the production of prekallikrein, the precursor to plasma kallikrein. A prior analysis from a Phase 1/2 trial showed that patients dosed with lonvo-z experienced an -89% reduction in plasma kallikrein sustained out to 36 weeks.
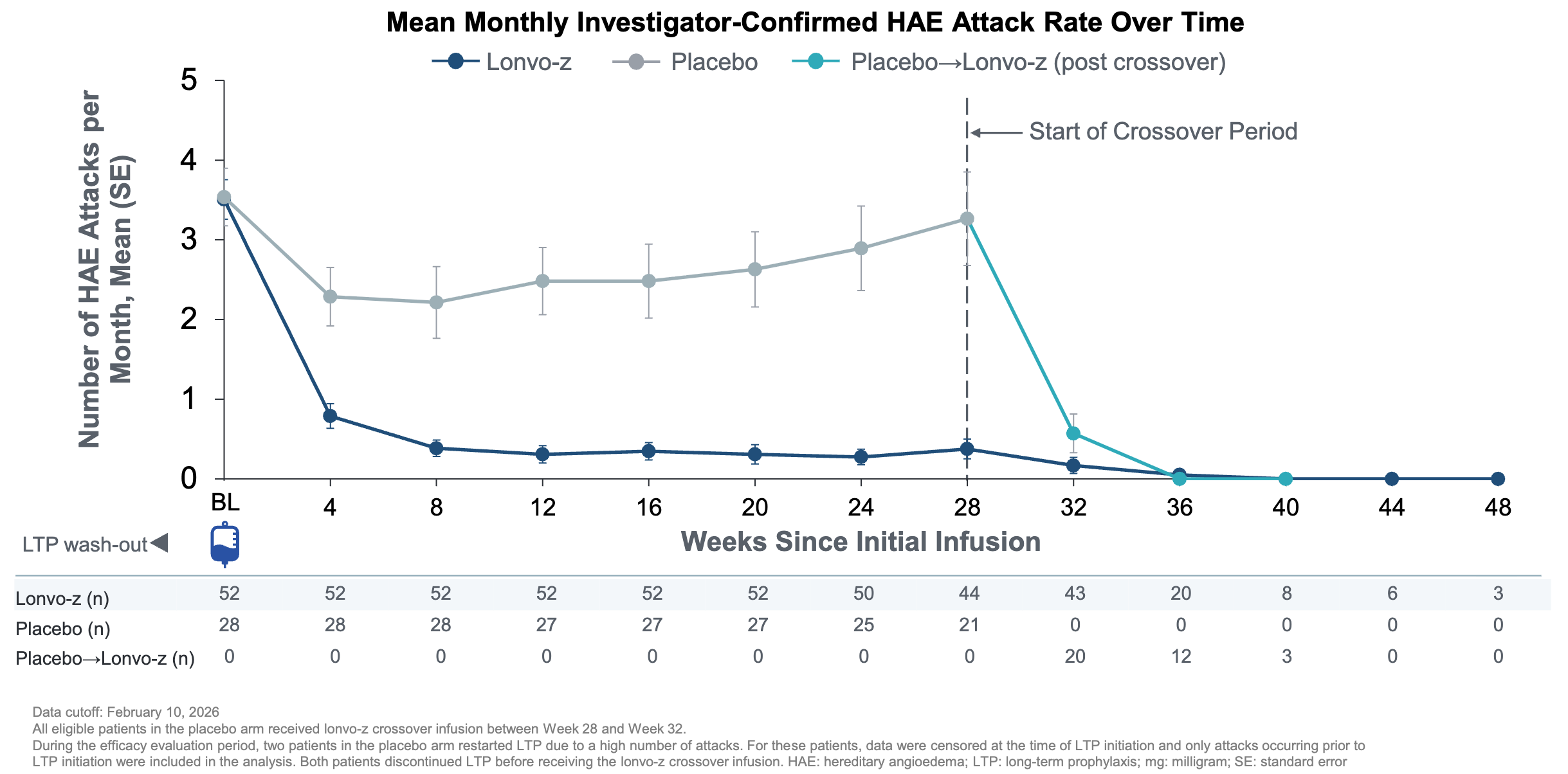
Trial design: The Phase 3 HAELO trial was a randomized, double-blind, placebo-controlled trial comparing 52 patients who received a one-time 50mg dose against 28 patients who received placebo. The trial recruited 80 adults and adolescents (16+) with Type I or II HAE. Patients were required to discontinue their baseline long-term prophylaxis (LTP) before dosing, forcing the therapy to demonstrate efficacy in a real-world state of vulnerability. The primary efficacy was measured between weeks 5 and 28 post-infusion.
Data (press release, slide deck): The trial met its primary endpoint (lonvo-z reduced attacks by 87% versus placebo between weeks 5 to 28, p<0.0001) and all key secondary endpoints with high statistical significance (attack fre rate 62% Lonvo-z versus 11% placebo, p<0.0001). As of the data cutoff, all patients who received lonvo-z at baseline or in crossover after week 28 remained LTP free. Furthermore, placebo patients who were crossed over to lonvo-z at week 28 saw a rapid reduction of HAE attacks (teal line in graph below). The most common adverse events were transient infusion-related reactions, headache, and fatigue. No Grade 3 or higher treatment-emergent events were reported.
Impact:
Reduced Treatment Burden: For the 62% who are attack-free, the need for bi-weekly subcutaneous injections (e.g., Takhzyro) or daily oral pills (e.g., Orladeyo) is eliminated.
Steady-State Control: Unlike traditional prophylaxis, which can have trough periods where breakthrough attacks occur, the genomic knockout provides a constant, non-fluctuating reduction in kallikrein.
Next steps: The trial sponsor has already initiated a rolling Biologics License Application (BLA) submission with the FDA as of late April 2026. The company expects to finish the BLA submission in the second half of 2026. If approved, the trial sponsor anticipates a U.S. launch in the first half of 2027. Patients in the HAELO trial will be followed for several years to monitor the long-term durability of the edit and potential delayed genomic risks. Additional clinical data from HAELO will be presented at the 2026 European Academy of Allergy and Clinical Immunology Congress (EAACI), taking place June 12-15 in Istanbul, Türkiye (abstract #100217).
Oruka / ORKA-001 (IL-23p19 mAb HLE) / Phase 2a (plaque psoriasis)
On April 27, 2026, Oruka Therapeutics announced positive interim Phase 2a results for ORKA-001, positioning it as a potential best-in-class therapy for moderate-to-severe plaque psoriasis due to its extreme durability and high efficacy.
Indication: The pathophysiology of plaque psoriasis is driven by a hyperactive IL-23/IL-17 immune axis, where IL-23 triggers Th17 cells to release cytokines like IL-17A and IL-17F, causing keratinocytes to proliferate rapidly and form raised, scaly plaques. The standard of care follows a severity-based escalation, increasingly moving toward high-efficacy systemic options for patients with moderate-to-severe disease. Topical therapies, including high-potency corticosteroids, Vitamin D analogs, and non-steroidal options like Vtama or Daliresp, remain the first line for localized disease. For patients who fail topicals or have extensive body surface area (BSA) involvement, the paradigm has shifted toward biologic agents that offer targeted inhibition of IL-23 (Skyrizi, Tremfya) or IL-17 (Bimzelx, Cosentyx), which can achieve complete or near-complete skin clearance (PASI 90/100). Additionally, the landscape has recently expanded to include advanced oral therapies, such as the TYK2 inhibitor Sotyktu and the newly approved oral IL-23 receptor antagonist Icotyde, providing patients with biologic-like efficacy in a convenient pill format.
Mechanism: ORKA-001 is a monoclonal antibody that targets the p19 subunit of interleukin-23 (IL-23), a key driver of the Th17 inflammatory pathway in psoriasis. It features proprietary Half-Life Extension (HLE) technology (YTE-like or similar Fc-engineering) designed to dramatically increase circulation time. A prior Phase 1 showed drug concentrations remained above effective trough levels for a full year after a single 600 mg dose, suggesting the potential for once- or twice-yearly maintenance dosing.
Trial design: The Phase 2a EVERLAST-A trial was a randomized, double-blind, placebo-controlled trial conducted in 84 adults with moderate-to-severe plaque psoriasis comparing 600 mg ORKA-001 at Week 0 and Week 4 (n=63) versus placebo (n=21). The primary endpoint was the percentage of patients reaching PASI 100 (complete skin clearance) at Week 16.
Data (press release, slide deck): The trial met its primary endpoint (16-week PASI 100 of 63.5% for ORKA-001 versus 4.8% placebo) and a key secondary endpoint (IGA 0/1 (Clear/Almost) 84.1% for ORKA-001 versus 4.8% for placebo). These results numerically outperformed historical benchmarks for currently approved therapies on a cross-trial basis (16-week PASI 100 of 63.5% for ORKA-001 versus 62% Bimzelx versus 43% Skyrizi versus 30% Icotyde; see graph below); however, cross-trial comparisons have inherent limitations and do not substitute for head-to-head studies.
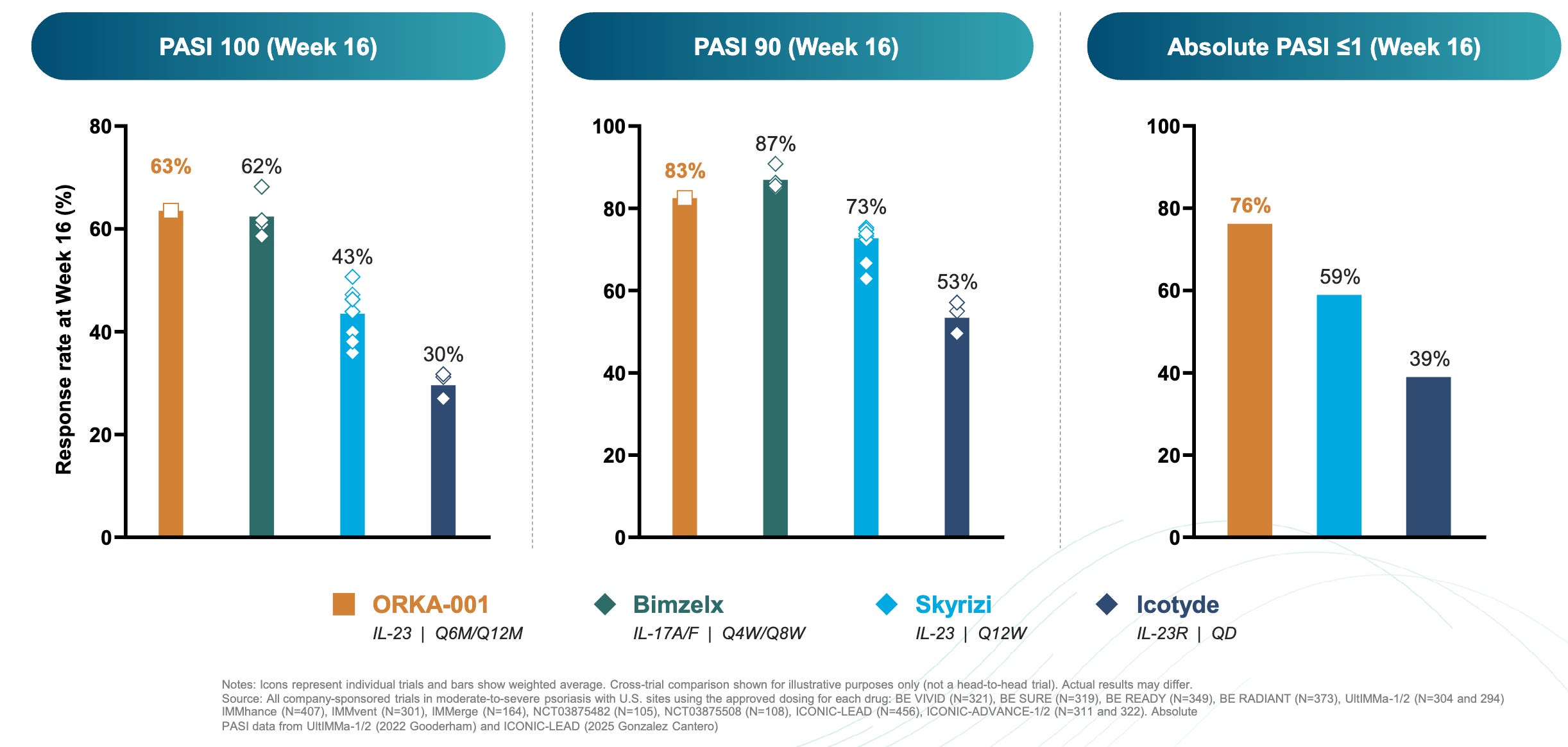
Impact:
Pushing the Efficacy Frontier: The 63.5% PASI 100 rate at 16 weeks is among the highest ever recorded for therapies in the plaque psoriasis space.
Convenient Dosing Paradigm: If once-yearly dosing is validated, it would represent a massive shift from the current standard of 4–6 injections per year, potentially improving patient adherence and quality of life.
Next steps: In 2H 2026, the trial sponsor expects to release of full EVERLAST-A data, including 28-week efficacy and 52-week follow-up for a subset of patients to confirm durability. A larger Phase 2b dose-ranging study is currently recruiting to identify the optimal induction and maintenance regimens. The trial sponsor expects to release results in 2027. Based on the Phase 2a strength, the company is preparing for Phase 3 initiation, likely following the conclusion of the Phase 2b dose-finding work.
Veradermics / VDPHL01 (ER oral minoxidil) / Phase 2/3 (androgenetic alopecia)
Indication: Androgenetic alopecia (AGA) is characterized by a genetically predetermined sensitivity of hair follicles to dihydrotestosterone (DHT), an androgen converted from testosterone by the enzyme 5-alpha reductase. This sensitivity triggers a process of follicular miniaturization, where the growth (anagen) phase progressively shortens and the hair follicles shrink, eventually producing fine, non-pigmented vellus hairs instead of thick terminal hairs. The current standard of care primarily targets these hormonal and physiological pathways through a combination of minoxidil, a vasodilator that prolongs the anagen phase and increases blood flow to the follicle, and finasteride, an oral 5-alpha reductase inhibitor that lowers systemic DHT levels. While these remain the primary FDA-approved interventions, the field is shifting toward optimized delivery systems, such as the extended-release oral minoxidil seen in VDPHL01, which aims to maximize the follicular conversion of minoxidil to its active sulfate form while mitigating the cardiovascular side effects associated with standard oral formulations.
Mechanism: VDPHL01 is an extended-release (ER) formulation of minoxidil, designed to solve the spike and crash problem of standard oral minoxidil (which was originally an antihypertensive). It uses a sustained-release matrix to maintain steady blood levels, avoiding the rapid plasma spikes that cause cardiac side effects like tachycardia. Minoxidil is a prodrug that must be converted to minoxidil sulfate by the enzyme SULT1A1 in the hair follicle. Standard oral tablets often saturate this enzyme’s capacity with a quick burst; the ER formulation provides a steady drip that optimizes this conversion, leading to better and more consistent hair growth.
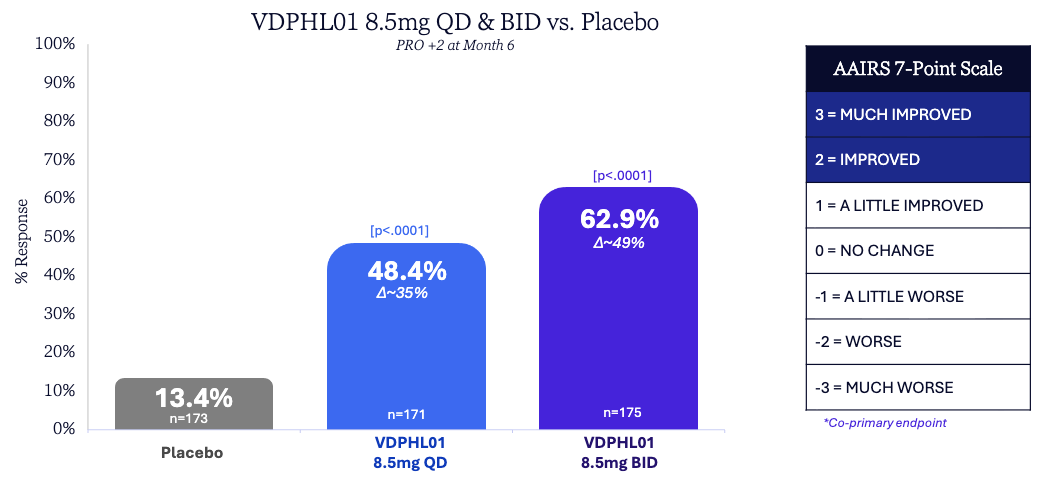
Trial design: The Phase 2/3 trial (Study ‘302’) randomized 519 men with mild-to-moderate pattern hair loss (Norwood stages II–V) to VDPHL01 8.5 mg Once-Daily (QD), VDPHL01 8.5 mg Twice-Daily (BID), or placebo for 6 months (primary endpoint) with a longer-term extension. The co-primary endpoints were Non-vellus Target Area Hair Count (TAHC) and patient-reported “improved” or “much improved” ratings on the AAIRS scale.
Data (press release, slide deck): The trial met all primary and key secondary endpoints with high statistical significance (see graphs below; p < 0.0001). tatistical separation from placebo was observed as early as Month 2, significantly faster than traditional topicals or orals. VDPHL01 was generally well-tolerated with overall AE rates similar to placebo. Most importantly, there were zero treatment-related serious adverse events (SAEs) and no cardiac events of special interest.
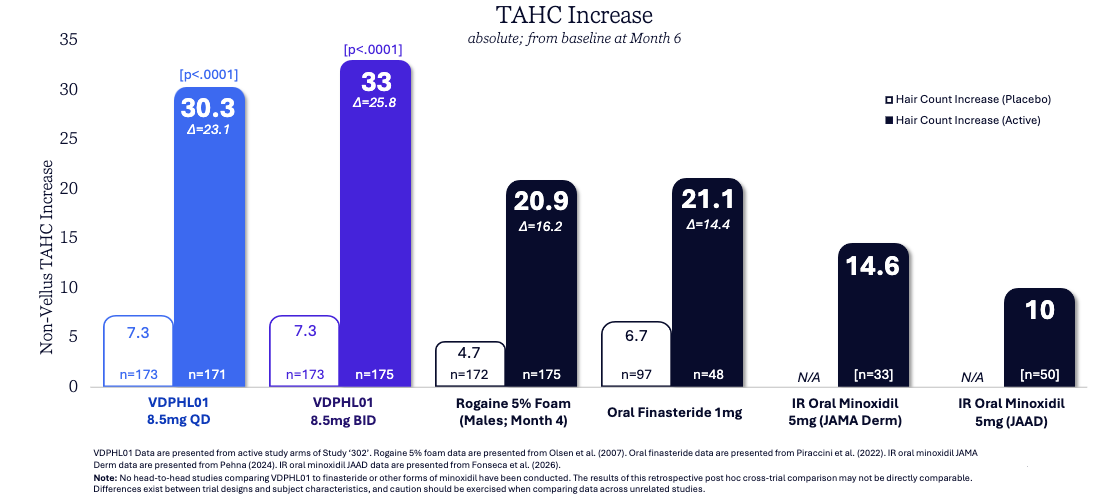
Impact:
The First Non-Hormonal Pill: If approved, it would be the first FDA-sanctioned oral non-hormonal treatment for hair loss in nearly 30 years, offering a potent alternative to finasteride (which carries sexual side-effect risks) and messy topicals.
Superior Potency: The growth of ~30–33 hairs/cm² numerically exceeds historical benchmarks for both topical minoxidil and oral finasteride on a cross-trial basis.
Potentially Improved Compliance: By eliminating the need for daily scalp applications and reducing the risk of heart palpitations, it targets a much higher patient adherence rate.
Next steps: The trial sponsor is running a second pivotal Phase 3 trial in men (Study ‘304’) and guides to topline results in the second half of 2026. An ongoing Phase 3 trial is evaluating the drug specifically for female pattern hair loss (Study ‘306’). The trial sponsor plans to submit a New Drug Application (NDA) to the FDA in late 2026 or early 2027, following the ‘304’ readout, positioning the drug for a potential 2027 launch.
Mirum / Brelovitug (anti-HBsAg mAb) / Phase 2b (Hepatitis D)
Indication: Hepatitis D (HDV) is a unique satellite virus that is obligate upon a co-existing Hepatitis B (HBV) infection, as it requires the Hepatitis B surface antigen (HBsAg) to wrap its own RNA genome and successfully infect hepatocytes. The pathophysiology is characterized by a high degree of viral replication that is more cytopathic and inflammatory than HBV alone, leading to rapid liver damage, necroinflammation, and a significantly accelerated progression to cirrhosis or hepatocellular carcinoma. The current standard of care has historically been limited to off-label use of pegylated interferon-alpha, which has a low success rate and significant side effects; however, the treatment landscape is shifting toward targeted therapies like bulevirtide (an entry inhibitor approved in Europe) and monoclonal antibodies like brelovitug that aim to disrupt the virus’s reliance on HBsAg, offering a more tolerable and effective means of achieving virologic suppression.
Mechanism: Brelovitug is a potent, pan-genotypic, fully human IgG1 monoclonal antibody targeting the Hepatitis B surface antigen (HBsAg). It binds to the antigenic loop of HBsAg, preventing HDV (which uses HBsAg as its envelope) from entering and infecting new hepatocytes. It facilitates the rapid removal of circulating HDV/HBV virions and subviral particles from the bloodstream. By depleting the HBsAg that typically exhausts the immune system, it may help restore antigen-specific T-cell function and anti-viral immunity.
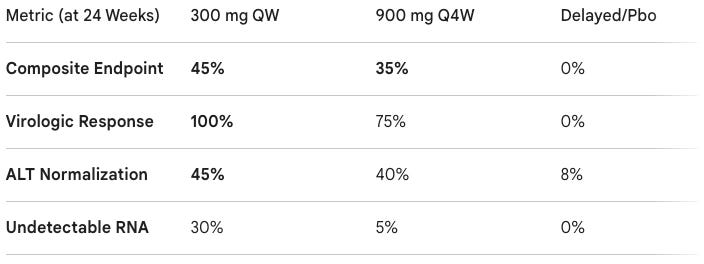
Trial design: The AZURE-1 Phase 2b trial randomized patients with chronic HDV, quantifiable HDV RNA, and elevated ALT to to 300 mg brelovitug once weekly (QW), 900 mg brelovitug once every 4 weeks (Q4W), or a delayed treatment (placebo) arm. The primary endpoint was A composite of virologic response (≥2log10 reduction in HDV RNA or undetectable) and ALT normalization at Week 24.
Data (press release): The study showed high statistical significance (p = 0.003 for the QW arm) compared to the control group. Brelovitug showed a “favorable profile” according to the trial sponsor with no serious treatment-related AEs or discontinuations. The most common events were mild injection-site reactions (14–20%) and musculoskeletal pain.
Impact:
First Effective Single-Agent: Hepatitis D is the most severe form of viral hepatitis, often leading to cirrhosis. Brelovitug’s ability to achieve 100% viral suppression as a monotherapy is a major breakthrough.
Tolerability Advantage: Current standard of care (interferons) has poor tolerability and high relapse rates. Brelovitug offers a potentially safer alternative.
Convenience Improvement: The success of the 900 mg monthly (Q4W) arm suggests that a simple monthly injection could be viable for long-term maintenance.
Next steps: The trial sponsor expects to present full results in a late-breaking poster session at the European Association for the Study of the Liver (EASL) congress in May 2026. The trial sponsor expects to report topline 24-week data from the pivotal Phase 3 studies, AZURE-1 and AZURE-4, by the end of 2026. If Phase 3 results confirm these findings, the trial sponsor plans to submit a BLA in 2027 under the FDA’s Breakthrough Therapy designation.
Incyte / Povorcitinib (JAK1 inhibitor) / Phase 3 (nonsegmental vitiligo)
Indication: Nonsegmental vitiligo is an autoimmune skin disorder characterized by the progressive destruction of melanocytes, primarily driven by a hyperactive IFN-γ / CXCL10 / CXCR3 signaling axis. In this pathway, CD8+ T cells infiltrate the skin and secrete interferon-gamma (IFN-γ), which triggers keratinocytes to produce chemokines that recruit even more autoreactive T cells, creating a self-sustaining feedback loop of pigment loss. The standard of care focuses on interrupting this immune attack and stimulating repigmentation, typically starting with topical corticosteroids or calcineurin inhibitors for localized disease. For patients with more extensive or recalcitrant involvement, treatment has been revolutionized by the approval of topical JAK inhibitors (such as ruxolitinib cream), which directly block the IFN-γ signaling loop, often used alongside narrowband UVB (NB-UVB) phototherapy to stabilize the disease and encourage the migration of melanocyte precursors from hair follicles to the skin’s surface.
Mechanism: Povorcitinib works by selectively inhibiting Janus kinase 1 (JAK1), a key signaling node in the autoimmune destruction of melanocytes. It interrupts the IFN-gamma / CXCL9 / CXCL10 signaling loop. In vitiligo, CD8+ T cells secrete IFN-gamma, which activates the JAK/STAT pathway in keratinocytes to recruit more T cells. By blocking this signal, povorcitinib halts the immune attack, allowing melanocyte precursors (stem cells in the hair follicle) to migrate back into the skin and restore pigment.
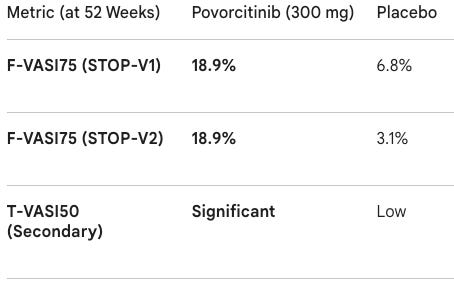
Trial design: The Phase 3 STOP-V1 & STOP-V2 trials randomized Over 900 adults with nonsegmental vitiligo involving ≥5% total body surface area (BSA) and ≥0.5% facial BSA to 300 mg povorcitinib once daily or placebo in the 52-week double-blind, placebo-controlled period. The primary endpoint was Facial Vitiligo Area Scoring Index 75 (F-VASI75) at Week 52.
Data (press release): The program achieved high statistical significance (p < 0.001) across both pivotal studies. The trials also met key secondary endpoints, including T-VASI50 (50% or more total body repigmentation), showing that the oral systemic approach works beyond just facial areas. The tolerability profile was consistent with previous JAK1 studies; the most common adverse events were acne, nasopharyngitis, and upper respiratory infections. No new safety signals were identified.
Impact:
Potentially Better Convenience: While Incyte’s Opzelura (ruxolitinib cream) is the standard for localized vitiligo, it is difficult to apply to large body areas. Povorcitinib offers a pill-once-daily solution for patients with extensive disease.
Efficacy Ceiling: Vitiligo repigmentation is a slow process. The 52-week data suggests that longer-term treatment leads to cumulative benefits, which is vital for a condition where patients often struggle with long-term adherence to topicals or phototherapy.
Next steps: The trial sponsor plans to present more detailed data from the Phase 3 program at a major medical congress in the second half of 2026. The trial sponsor expects to file a supplemental New Drug Application (sNDA) for nonsegmental vitiligo in the first half of 2027. Povorcitinib is also in late-stage development for hidradenitis suppurativa (HS) and prurigo nodularis, with the HS application already under FDA review and a potential launch in early 2027, per the trial sponsor’s guidance.
Descriptive data releases without numerical data
Pfizer / Elrexfio (BCMA x CD3 TCE) / P3 success (2L MM): On April 29, 2026, Pfizer announced that the Phase 3 MagnetisMM-5 trial of Elrexfio (elranatamab) met its primary endpoint, demonstrating a statistically significant and clinically meaningful improvement in progression-free survival (PFS) as a monotherapy compared to the standard-of-care triplet (daratumumab, pomalidomide, and dexamethasone) in patients with relapsed or refractory multiple myeloma (RRMM) who have received at least one prior line of therapy. Elrexfio is a BCMA x CD3 bispecific T-cell engager (TCE) that functions by simultaneously binding to B-cell maturation antigen (BCMA) on myeloma cells and the CD3 receptor on T cells, thereby bridging them and inducing potent T-cell-mediated cytotoxicity. The trial’s interim results, which exceeded the pre-specified hazard ratio threshold for efficacy, support moving the therapy into second-line (2L) treatment—significantly earlier than its current fourth-line accelerated approval—and reinforce a manageable safety profile consistent with prior data, featuring no new safety signals. Following this success, the trial sponsor plans to present detailed findings at an upcoming medical congress and engage with global regulatory authorities for label expansion, while continuing to evaluate overall survival (OS) as a key secondary endpoint and progressing with other MagnetisMM trials exploring combination regimens in earlier settings.
To contact us, please send us an email at biotechreadout@gmail.com
Disclaimers
Investigational Status Disclaimer
The therapeutic candidates discussed in this newsletter are currently in clinical development and have not been approved for commercial sale by the U.S. Food and Drug Administration (FDA), the European Medicines Agency (EMA), or other global regulatory authorities. Their safety and efficacy have not been established. References to pipeline products and ongoing clinical trials involve significant risks and uncertainties. Statements regarding the potential safety, potency, or efficacy of investigational drugs reflect current hypotheses and are not a guarantee of future performance or regulatory clearance. The outcome of clinical trials is inherently unpredictable, and clinical results from earlier stages may not be predictive of results in later, larger-scale trials.
No Medical Advice Disclaimer
This newsletter is for informational and educational purposes only. The content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or another qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read in this publication.
No Patient-Provider Relationship Disclaimer
The information provided in this newsletter is for educational and analytical purposes only. Receipt of this information, or any interaction with this content, does not create a physician-patient, pharmacist-patient, or any other professional-provider relationship between you and the authors or publishers. This newsletter should not be used as a substitute for a personal consultation with a qualified healthcare professional.
Forward-Looking Statements Disclaimer
This newsletter contains “forward-looking statements” regarding future events, including clinical trial timing, regulatory milestones, and projected market performance. These statements are based on current expectations and assumptions that are subject to significant risks and uncertainties. Actual results may differ materially from those expressed or implied. We undertake no obligation to update these statements as a result of new information or future developments.
Third-Party Links & Content Disclaimer
This newsletter contains links to third-party websites, including clinical trial registries and corporate presentations. Biotech Readout does not endorse, guarantee, or assume responsibility for the accuracy or reliability of any information offered by third-party providers.
Errors and Omissions Disclaimer
While we strive for technical accuracy, the information in this newsletter is provided on an “as is” basis with no guarantees of completeness, accuracy, or timeliness. Biotech Readout assumes no liability for any errors or omissions in the content of this publication.
Non-Endorsement Disclaimer
Any reference to specific commercial products, processes, or services by trade name, trademark, or manufacturer does not constitute or imply an endorsement or recommendation by the author. All trademarks are the property of their respective owners.
No Investment Advice Disclaimer
This newsletter is for informational purposes only and does not constitute financial, investment, or legal advice. The author is not a registered investment advisor. You should consult with a professional financial advisor before making any investment decisions. The biotechnology sector is highly volatile; past performance is not indicative of future results.
Conflict of Interest Disclaimer
The author of this newsletter maintains a position of independence. At the time of publication, the author holds no direct financial interest, equity, or options in any of the companies mentioned in this report. No compensation has been received from any third party to feature or analyze specific therapeutic candidates or corporate entities.