
First approval for genetic hearing loss, AACR and AAN data and more
Weekly Readout #4: Week of April 20, 2026

Disclaimer: This newsletter is for educational and informational purposes only and does not constitute medical, investment, or financial advice, nor does it establish a provider-patient relationship. Content may include forward-looking statements and discussions of investigational therapeutic candidates that are not FDA/EMA approved; their safety and efficacy remain unestablished and clinical outcomes are unpredictable. While we strive for accuracy, all information is provided as is without guarantees. This newsletter is independent, and the author holds no financial positions in the companies mentioned nor receives third-party compensation for this coverage. Please find a complete version of our disclaimers at the bottom of this article and on our About page.

Press ⬇️ to go down to a section and ⬆️ to go back up to the Table of Contents
Table of Contents
This week, we discuss:
Regeneron / Otarmeni (AAV1-OTOF) / FDA-approval (severe-to-profound/profound OTOF-related hearing loss); first FDA-approved gene therapy for genetic hearing loss ⬇️
Monopar / ALXN1840 (bis-choline tetrathiomolybdate) / Phase 3 (Wilson disease) ⬇️
GSK / Risvutatug rezetecan (B7-H3 ADC) / Phase 1 (non-squamous NSCLC) ⬇️
Merck / MK-2010 (PD1 x VEGF bsAb) / Phase 1 (NSCLC) ⬇️
Nektar / Rezpeg (IL-2 pathway agonist) / Phase 2b OL (alopecia areata) ⬇️
Johnson & Johnson / Carvykti (BCMA CAR-T) / Phase 2 (smoldering MM) ⬇️
AstraZeneca / Tozorakimab (anti-IL33 mAb) / Phase 3 (COPD) ⬇️
Regeneron / Otarmeni (AAV1-OTOF) / FDA-approval (severe-to-profound/profound OTOF-related hearing loss)
On April 23, 2026, the FDA granted accelerated approval to Otarmeni (lunsotogene parvec-cwha) via CNPV, marking the first FDA-approved example of a gene therapy to restore a neurosensory function to normal levels. It is indicated for pediatric and adult patients with severe-to-profound hearing loss (any frequency >90 dB HL) associated with molecularly confirmed biallelic variants in the OTOF gene, preserved outer hair cell function, and no prior cochlear implant in the same ear. Importantly, the manufacturer (Regeneron) will provide Otarmeni for free to clinically eligible individuals in the U.S.
Indication: The OTOF gene, which encodes the protein otoferlin. Otoferlin acts as a “calcium sensor” at the synapse between inner hair cells and the auditory nerve; without it, sound signals are never transmitted to the brain. The clinical standard of care has historically focused on bypassing the biological synaptic failure through cochlear implantation (CI). Because the auditory nerve is typically healthy in these patients, electrical stimulation from a CI can effectively transmit sound signals to the brain, and OTOF patients often demonstrate superior speech perception outcomes compared to those with other genetic forms of deafness.
Mechanism: Otarmeni uses a dual-adeno-associated virus (AAV1) vector system. Because the OTOF gene is too large for a single AAV, it is split into two halves that recombine within the cell to form the full-length functional gene. Expression is driven by a proprietary Myo15 cell-specific promoter, ensuring otoferlin is only produced in the inner hair cells where it is naturally required. A single, one-time intracochlear infusion performed surgically under general anesthesia, similar to the approach used for cochlear implantation.
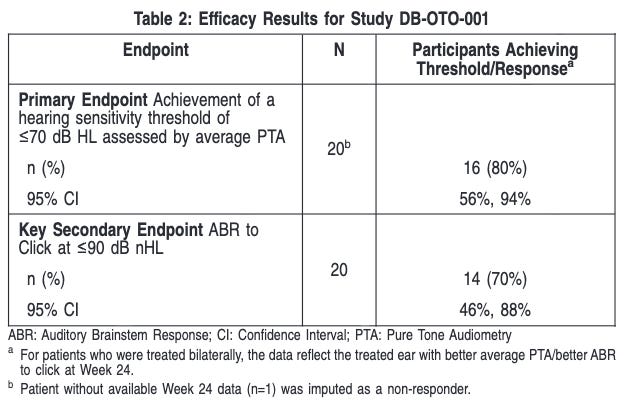
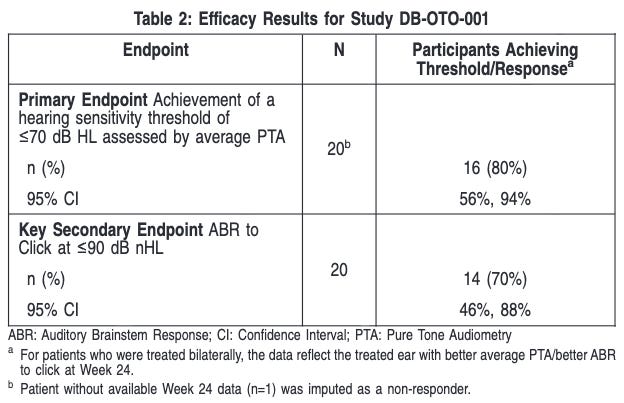
Trial Design: Accelerated approval was based on the CHORD trial, an ongoing, registrational, multi-center, Phase 1/2 open-label study. The trial included 20 children and adolescents (aged 10 months to 16 years) with molecularly confirmed biallelic OTOF variants. Patients received either a unilateral (one ear, n=10) or bilateral (both ears, n=10) infusion. The primary efficacy endpoint was the achievement of a hearing sensitivity threshold 70 dB HL assessed by average pure tone audiometry (i.e., average of PTA thresholds at 0.5, 1.0, 2.0, and 4.0 kHz) at Week 24 after product administration.
Data: At Week 24, a total of 9/20 (45%) patients achieved an average PTA threshold 45 dB HL (ability to hear soft conversational speech level) and 3/20 (15%) achieved 25 dB HL (normal hearing level i.e., ability to hear whispers). One patient received a cochlear implant as rescue treatment approximately 8 months after Otarmeni administration in the same ear following determination of treatment failure. Among the 20 patients, 12 patients were evaluated at Week 48 after product administration. Of these, all 9 patients who previously had a response at Week 24 and who were evaluated at Week 48 maintained their response. One additional patient who had not initially achieved response at Week 24, achieved an average PTA threshold 70 dB HL by Week 48, resulting in a total of 10/12 (83%) patients who achieved an average PTA threshold 70 dB HL by Week 48. Eight of the 12 (67%) patients achieved an average PTA threshold 45 dB HL and 5 of the 12 patients (42%) achieved 25 dB HL by Week 48. Additionally, 9 of the 12 patients (75%) demonstrated presence of auditory brainstem response (ABR) to a click (broadband sound) stimulus of 90 dB nHL by Week 48. The most common adverse events included otitis media (38%), vomiting (33%), nausea (29%), dizziness (21%), procedural pain (17%), gait disturbance (8%), and nystagmus (8%). No serious drug-related adverse events were reported.
FDA label for Otarmeni Impact:
Restoration vs. Amplification: Unlike cochlear implants, which bypass the hair cells to stimulate the nerve electrically, Otarmeni restores the natural biological pathway, potentially allowing for better sound quality, music appreciation, and speech development.
Economic Accessibility: Regeneron has committed to providing the drug at no cost to clinically eligible patients in the U.S. (though surgical/facility costs may still apply).
Regulatory Milestone: This was one of the fastest BLA approvals in FDA history (61 days) and the first under the National Priority Voucher program.
Next Steps: As an accelerated approval, the trial sponsor must verify clinical benefit through the confirmatory portion of the CHORD trial, which will focus on long-term durability and speech perception outcomes. Participants will be followed for several years to ensure the AAV expression remains stable and to monitor for any delayed immune responses. The trial sponsor anticipates regulatory filings in the EU (EMA) and other major markets throughout 2026.
Monopar / ALXN1840 (bis-choline tetrathiomolybdate) / Phase 3 (Wilson disease)
ALXN1840 (tiomolibdate choline), a late-stage therapy for Wilson disease, has shown significant potential in addressing the neurological symptoms that often persist or worsen with existing treatments. Following recent data presentations at the American Academy of Neurology (AAN) 2026, here is the summary of its current clinical standing.
Indication: Wilson disease is a rare autosomal recessive disorder characterized by the body’s inability to eliminate copper, leading to toxic accumulation in the liver, brain, and other vital organs. The disease is caused by mutations in the ATP7B gene, which encodes a transmembrane protein responsible for transporting copper into the bile for excretion and incorporating it into ceruloplasmin. When ATP7B is defective, copper cannot be excreted into the bile. It builds up in hepatocytes (liver cells). Once the liver’s storage capacity is exceeded, copper enters the bloodstream as “free” (non-ceruloplasmin-bound) copper. This generates reactive oxygen species through the Fenton reaction, leading to lipid peroxidation and cell death. The excess copper deposits in secondary tissues including the CNS (causing tremors, dystonia, and psychiatric symptoms), eyes (causing rings in the cornea called Kayser-Fleischer rings), and blood (causing Coombs-negative hemolytic anemia). Standard of care involves lifelong treatment with copper chelators and zinc salts as well as dietary changes to reduce copper levels.
Mechanism: ALXN1840 is a first-in-class selective copper-binding agent. It forms a highly stable tripartite complex with copper and albumin in the blood and liver. By sequestering excess copper into these complexes, it renders the copper redox-inactive (preventing oxidative damage) and promotes its excretion through the biliary tract, the body’s natural route, rather than the kidneys. Unlike standard chelators, it is designed to mobilize copper rapidly without causing the initial neurological spike often seen when traditional treatments mobilize copper into the bloodstream and brain.
Trial design: The FoCus trial (NCT03403205) was a randomized, rater-blinded, multi-center study comparing ALXN1840 to Standard of Care (SoC) such as penicillamine, trientine, or zinc. Patients included both treatment-naïve and treatment-experienced (previously on SoC for at least 1 year) patients aged 12 and older.
Data: The latest readout emphasized “clinically meaningful” benefits for patients including lower rates of Neurologic Worsening (9% vs 25% SOC, p=0.038) and higher rates of Neurologic Improvement (45% vs 32% SOC), CGI-S (61% vs 17% SOC, p=0.008), and CGI-I (47% vs 19% SOC, p=0.003). Benefits were sustained and continued to increase over approximately 3 years of long-term follow-up. The profile was favorable across 266 patients. Drug-related Serious Adverse Events (SAEs) occurred in only 4.9% of patients, with neurologic SAEs in < 1%. No treatment-related deaths were reported.
Impact: Wilson disease management has historically been plagued by “iatrogenic worsening,” where starting treatment causes a sudden release of copper that permanently damages the brain. ALXN1840’s data suggests it could:
Reduce Permanent Disability: By significantly lowering the rate of worsening (9% vs 25%).
Simplify Dosing: Offers a potential once-daily oral option with better tolerability than traditional heavy-metal chelators.
Next steps: The trial sponsor plans to submit a New Drug Application (NDA) to the FDA in mid-2026. The company recently appointed a Chief Commercial Officer to build the launch framework. Following the U.S. filing, regulatory submissions in other global markets (EMA/MHRA) are expected to follow.
GSK / Risvutatug rezetecan (B7-H3 ADC) / Phase 1 (non-squamous NSCLC)
Data from the AACR 2026 annual meeting (presented April 2026) highlighted the potent activity of risvutatug rezetecan (formerly HS-20093/GSK5764227) in combination with the PD-L1 inhibitor adebrelimab. This combination is being investigated as a second-line (2L) treatment in patients with non-squamous non-small cell lung cancer (nsq-NSCLC).
Indication: Non-squamous non-small cell lung cancer (nsq-NSCLC) is the most common subtype of lung cancer, primarily comprising adenocarcinomas. Unlike squamous cell carcinoma, it typically originates in the peripheral lung tissues and is frequently associated with specific genetic “driver” mutations. The development of nsq-NSCLC involves a complex interplay of genetic mutations and environmental factors (e.g., smoking, radon, asbestos). Most nsq-NSCLC arises from Type II alveolar cells, which are responsible for surfactant production and lung repair. Nsq-NSCLC is characterized by a high frequency of actionable mutations, including EGFR (approx. 15-20% in Western populations), KRAS (approx. 30%), ALK, ROS1, and MET. Tumors often express PD-L1, a protein that suppresses the immune response, allowing cancer cells to evade T-cell detection. Survival is heavily dependent on the stage at diagnosis (5-year survival 62-73% for localized Stage I/II, 35-47% for regional Stage III, and 8-12% for Stage IV with brain/bone/liver metastasis). While rates have improved significantly due to targeted therapies and immunotherapy, metastatic disease remains a challenge. The current standard of care for advanced non-squamous NSCLC is centered on a biomarker-first approach, where treatment is dictated by the presence of actionable genetic drivers or PD-L1 expression levels. For patients without specific mutations (like EGFR or ALK), the backbone of frontline therapy typically involves platinum-based chemotherapy combined with PD-(L)1 immunotherapy, such as the KEYNOTE-189 regimen of Keytruda, carboplatin, and pemetrexed. If a patient progresses on this frontline treatment, the historical standard has been docetaxel (with or without ramucirumab), though this second-line setting is where newer assets like risvutatug rezetecan are currently challenging the status quo due to the relatively limited efficacy and high toxicity of traditional taxane chemotherapy.
Mechanism: Risvutatug rezetecan is a next-generation B7-H3-directed antibody-drug conjugate (ADC). B7-H3 (CD276) is an immune checkpoint molecule overexpressed in various solid tumors, including NSCLC, and is associated with poor prognosis and immune evasion. Risvutatug rezetecan utilizes a topoisomerase I inhibitor (exatecan derivative) payload. The combination with adebrelimab (a PD-L1 monoclonal antibody) aims to leverage the “immunogenic cell death” induced by the ADC’s cytotoxic payload, which can sensitize the tumor microenvironment to checkpoint inhibition, even in patients who previously progressed on immunotherapy.
Trial design: The data comes from a multi-center, open-label Phase 1 expansion cohort conducted in China (Hansoh Pharma). It enrolled patients with advanced non-squamous NSCLC without actionable genomic alterations (e.g., EGFR/ALK negative) who had failed at least one prior line of therapy (typically platinum-based chemo + IO).
Data: The combination demonstrated encouraging efficacy signals in 2L NSCLC data to date including 47.1% ORR, 14 months mPFS, and 85% DCR in the efficacy-evaluable set (n=34) with a data cutoff of January 20, 2026.
Impact:
Potentially redefining 2L Standard: If these results hold in larger trials, this combination could displace docetaxel as the preferred second-line treatment for nsq-NSCLC.
Overcoming IO Resistance: The high ORR in a post-PD-1/L1 population suggests that B7-H3 targeting effectively bypasses or reverses common mechanisms of resistance to traditional immunotherapy.
Next steps: The trial sponsor is initiating/expanding global Phase 3 trials (e.g., the ARTEMIS program) to validate these China-specific results in a broader Western population. Based on the Breakthrough Therapy Designation already held for other indications (SCLC), the NSCLC data likely sets the stage for an expedited regulatory pathway. Parallel efforts are ongoing in Small-Cell Lung Cancer (SCLC), where risvutatug rezetecan recently received Orphan Drug Designation in Japan (March 2026).
Merck / MK-2010 (PD1 x VEGF bsAb) / Phase 1 (NSCLC)
Data for MK-2010 (also known as LM-299), a PD-1 x VEGF bispecific antibody, was a key highlight at the AACR 2026 annual meeting (April 2026). Merck acquired this asset from LaNova Medicines (now a subsidiary of Sino Biopharm) in late 2024 to bolster its post-Keytruda strategy.
Indication: Non-small cell lung cancer (NSCLC) arises from the malignant transformation of bronchial epithelial cells, driven by the accumulation of genetic alterations, such as mutations in KRAS, EGFR, or ALK, and environmental insults like tobacco smoke that disrupt normal cell signaling and apoptosis. These changes lead to uncontrolled cellular proliferation and the remodeling of the tumor microenvironment to support angiogenesis and immune evasion, often through the upregulation of the PD-1/PD-L1 pathway. The current standard of care is heavily dictated by molecular profiling and disease stage; early-stage tumors are primarily managed with surgical resection often followed by adjuvant chemotherapy or immunotherapy, while advanced or metastatic disease requires a biomarker-first strategy. For patients with actionable mutations, frontline therapy involves targeted tyrosine kinase inhibitors (TKIs), whereas those without such drivers typically receive a combination of platinum-doublet chemotherapy and immune checkpoint inhibitors (anti-PD-1/PD-L1). As the disease progresses, treatment shifts toward subsequent lines of therapy, such as docetaxel or increasingly prevalent antibody-drug conjugates (ADCs) and bispecific antibodies, to overcome acquired resistance.
Mechanism: MK-2010 is a next-generation bispecific antibody (bsAb) designed to simultaneously hit two proven pathways. Dual Blockade: It inhibits the PD-1/PD-L1 checkpoint (releasing the “brakes” on T-cells) and the VEGF pathway (inhibiting tumor angiogenesis and normalizing the tumor vasculature). It utilizes a differentiated 2+1 or similar structure (an anti-VEGF antibody linked to two single-domain anti-PD-1 antibodies). This is intended to increase binding avidity in the presence of both targets, potentially improving selectivity for the tumor microenvironment over healthy tissue.
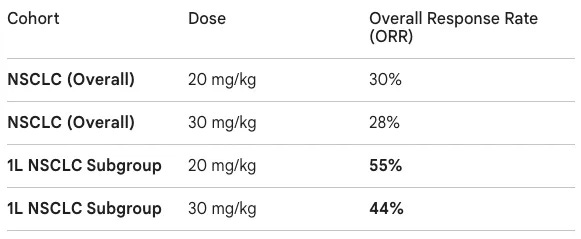
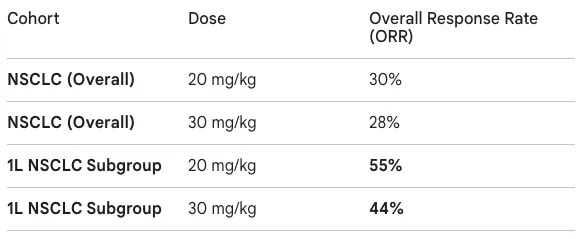
Trial design: The data presented at AACR 2026 came from a Phase 1 study primarily conducted in China. The trial recruited patients with advanced solid tumors, with a heavy emphasis on a backfill cohort of 72 patients with NSCLC. NSCLC patients were required to have PD-L1 expression (TPS ≥1%) and no actionable genomic alterations (EGFR/ALK negative). The study explored doses of 20 mg/kg (low dose) and 30 mg/kg (high dose) administered intravenously every three weeks (Q3W).
Data: The readout provided a competitive first look at MK-2010’s activity compared to rivals like ivonescimab. In the initial low-dose expansion, 6 out of 11 patients responded; in the high-dose expansion, 4 out of 9 patients responded. While the median follow-up was relatively short (~3.3 months), the responses in the frontline (1L) setting were particularly notable. The 1L ORR of 55% was specifically for the 20 mg/kg dose; the 30 mg/kg dose showed a slightly lower 44% ORR in the same setting. Safety was generally consistent with the PD-1 and VEGF classes. Grade 3/4 Treatment-Emergent Adverse Events (TEAEs) occurred in 17% (low dose) and 27% (high dose). Common VEGF-related AEs included hypertension (~9%) and proteinuria (~5%).
Impact:
The “Ivonescimab” Challenger: Following the HARMONi-2 data showing ivonescimab’s superiority over Keytruda, MK-2010 is a countermeasure by Merck to maintain their competitive advantage in the lung cancer market.
Frontline Potential: The 55% ORR in the 1L setting suggests that combining these two mechanisms into a single molecule may be superior to the historical attempts at combining two separate monoclonal antibodies.
Next steps: The trial sponsor is finalizing the Recommended Phase 2 Dose (RP2D), likely settling on the 20 mg/kg or 30 mg/kg Q3W schedule. They are focused on transitioning from the China-based Phase 1 to global Phase 2/3 trials. Furthermore, Merck is exploring pairing MK-2010 with its own internal assets, such as the TROP2 ADC (sacituzumab tirumotecan), to further move the needle in PD-L1 low or refractory populations.
Nektar / Rezpeg (IL-2 pathway agonist) / Phase 2b OL (alopecia areata)
Data from the REZOLVE-AA Phase 2b trial (presented April 20, 2026) highlights a unique “slow-burn” efficacy profile for rezpegaldesleukin (Rezpeg). While the trial initially missed its primary endpoint in the randomized controlled portion, the 52-week data suggests that its immune-modulating mechanism requires a longer induction period than traditional therapies.
Indication: Alopecia areata is an autoimmune condition characterized by the sudden loss of immune privilege in the hair follicle, where a breakdown in self-tolerance leads CD8+ T cells to attack the hair bulb. This inflammatory process, primarily driven by the JAK-STAT signaling pathway and cytokines like IFN-gamma and IL-15, forces follicles into a premature telogen (resting) phase without permanently destroying the follicle’s regenerative potential. The standard of care is stratified by the extent of hair loss; localized patches are typically managed with intralesional corticosteroids (e.g., triamcinolone) or high-potency topical steroids to suppress regional inflammation. For patients with severe or extensive disease (alopecia totalis or universalis), treatment has shifted toward systemic therapy with JAK inhibitors, such as baricitinib or ritlecitinib, which provide broad suppression of the inflammatory signaling cascades. Other options include topical immunotherapy (e.g., DPCP) to induce a diversionary allergic contact dermatitis, though the field is increasingly moving toward targeted biologics, like the IL-2 pathway agonists you mentioned, to restore long-term immune homeostasis.
Mechanism: Rezpeg is a first-in-class IL-2 receptor pathway agonist. It is designed to selectively stimulate and expand regulatory T cells (Tregs) by binding to the IL-2 receptor complex. In alopecia areata (AA), the loss of immune privilege at the hair follicle leads to T-cell-mediated destruction. Rezpeg aims to restore immune homeostasis, slowing down the autoimmune attack, rather than broadly suppressing the immune system like JAK inhibitors.
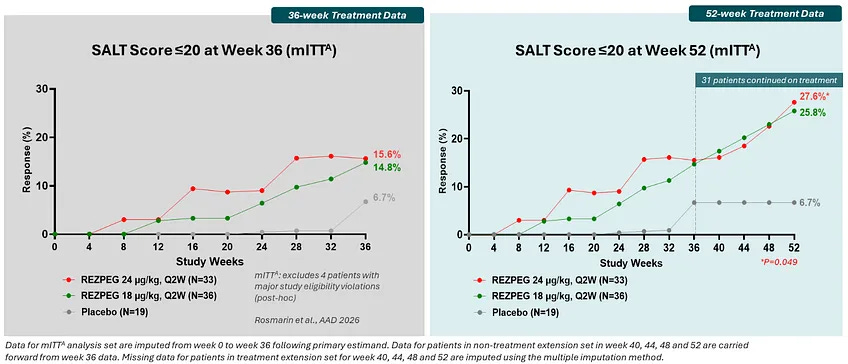
Trial design: The Phase 2b REZOLVE-AA study was a randomized, double-blind, placebo-controlled trial. It included 92 adults with severe-to-very-severe alopecia areata (≥50% scalp loss). In the initial phase (36 weeks), patients were randomized to low-dose (18 µg/kg), high-dose (24 µg/kg), or placebo twice monthly. in the extension phase (Weeks 36–52), patients who demonstrated growth but still had a SALT >20 were allowed to continue on a blinded extension to evaluate the durability and deepening of response.
Data: The trial presents a tale of two readouts. The trial technically missed its primary endpoint (mean change in SALT score) at Week 36 in the initial phase (grey box in figure below). The trial did not meet its primary endpoint in the intent-to-treat population, but a post-hoc, exploratory analysis excluding four patients suggested nominal significance, though such analyses are hypothesis-generating and require validation in future studies. The extended treatment showed that responses continued to mature significantly over time (green box in figure below). In the 52-week data, the SALT≤20 rates were 25.8% (low dose) and 27.6% (high dose). The 52-week data demonstrated a deepening of response, with 94% of patients completing the extension phase without any discontinuations due to adverse events. The safety profile remained favorable with no new signals or discontinuations during the 16-week extension period; the most common events were mild-to-moderate injection site reactions.
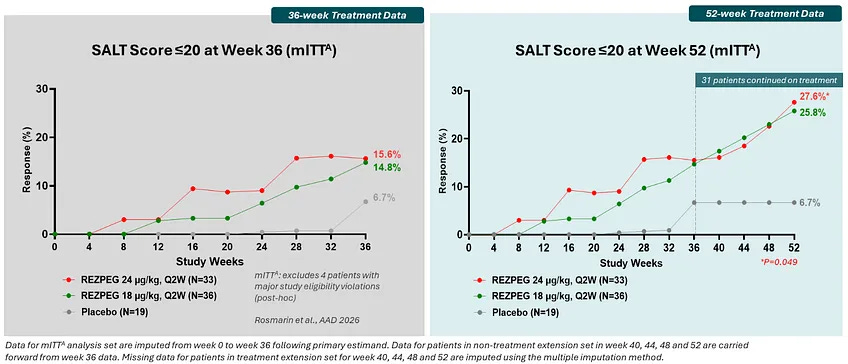
Source: Nektar presentation, slide 14 Impact:
The First Biologic Alternative: If approved, Rezpeg would be the first biologic for AA, providing an alternative to JAK inhibitors (e.g., baricitinib, ritlecitinib) which carry boxed warnings for serious infections, malignancy, and MACE.
Homeostatic vs. Suppressive: Unlike JAKs, which require continuous suppression, Rezpeg’s Treg-mediated approach offers the potential for long-term disease control and possibly “treatment-free” intervals if immune tolerance is successfully re-established.
Next steps: The trial sponsor has indicated that the 52-week data clarifies the need for a longer induction period (likely 52 weeks instead of 36) in registrational trials. The company plans to meet with the FDA in 2026 to finalize the Phase 3 design based on these results. Industry analysis are watching for off-treatment data to see if the hair growth is maintained after dosing stops, a key differentiator for the Treg mechanism.
Johnson & Johnson / Carvykti (BCMA CAR-T) / Phase 2 (smoldering MM)
Data presented at the AACR 2026 annual meeting (April 2026) by Dr. Omar Nadeem from the Dana-Farber Cancer Institute showcased the potential for early interception in smoldering multiple myeloma using Carvykti. The data were concurrently published in Nature Medicine.
Indication: Smoldering Multiple Myeloma (SMM) is an intermediate, asymptomatic clinical stage of plasma cell dyscrasia characterized by a clonal proliferation of malignant plasma cells in the bone marrow. The pathophysiology involves a “two-hit” genetic model: the initial progression from monoclonal gammopathy of undetermined significance (MGUS) is often driven by primary translocations (e.g., involving the IGH locus on chromosome 14) or hyperdiploidy. This creates a stable but abnormal clone. The progression to symptomatic disease is then triggered by secondary genetic hits, such as mutations in KRAS, NRAS, or MYC, alongside a permissive bone marrow microenvironment that fails to suppress the malignant expansion, eventually leading to end-organ damage. The standard of care for SMM is currently transitioning from a “watchful waiting” approach to more proactive risk stratification. For low-to-intermediate risk patients, the consensus remains active observation with serial monitoring of laboratory markers every 3 to 6 months to detect early signs of progression. However, for high-risk SMM (defined by the “20/2-20” criteria: ≥20% bone marrow plasma cells, M-protein >2 g/dL, and an involved/uninvolved free light chain ratio >20), clinical guidelines and recent trial data increasingly support early intervention. Treatment in these cases typically involves lenalidomide, either alone or in combination with dexamethasone, to delay the onset of symptomatic multiple myeloma and improve progression-free survival.
Mechanism: Carvykti is a BCMA-directed CAR-T cell therapy that utilizes a dual-epitope binding design to target B-cell maturation antigen (BCMA) on plasma cells. The rationale for using it in smoldering multiple myeloma (SMM) is that T cells in these patients are “fitter” and less exhausted than those in heavily pre-treated patients, potentially leading to more robust expansion and durability. It aims to eradicate the precursor malignant clones before they cause end-organ damage (CRAB criteria: calcium elevation, renal insufficiency, anemia, bone lesions).
Trial design: The CAR-PRISM trial (NCT05767359) is an investigator-initiated Phase 2 study evaluating a single infusion of Carvykti without induction or bridging therapy. The trial involved 20 patients with high-risk SMM, defined by the 20/2/20 criteria (BM plasma cells >20%, M-protein >2 g/dL, and FLC ratio >20). Primary endpoints included safety and dose-limiting toxicities; secondary endpoints included Overall Response Rate (ORR) and Minimal Residual Disease (MRD) negativity.
Data: The results demonstrated high rates of MRD negativity in this small study population; 100% of patients (20/20) achieved MRD negativity (10-6 sensitivity) within 2 months. At a median follow-up of 15.3 months, all patients remained MRD-negative. Among 16 patients with ≥6 months follow-up, the Complete Response (CR) rate was 100%. No instances of disease progression or deaths were observed during the follow-up period. While low-grade cytokine release syndrome (CRS) was universal (100% Grade 1/2), classic ICANS was managed well. 7 cases of non-ICANS neurologic toxicities (NINTs) occurred, including facial nerve palsy, which is a known but manageable side effect for BCMA CAR-Ts. This included 4 cases of facial nerve palsy (all resolved) and 3 cases of mild motor symptoms that remained improved but present at last follow-up.
Impact:
Potential Shift in Paradigm: Currently, SMM is managed with “watchful waiting” or mild intervention (Dara-Faspro). This trial suggests that Carvykti could offer the potential for long-term disease-free survival in high-risk patients.
Immune Robustness: The data confirmed that earlier intervention leads to rapid and deeper responses compared to later lines of therapy.
Next steps: The primary focus of the Phase 2 trial is determining if these MRD-negative responses can be sustained for 5+ years, which would define a functional cure. Based on these pilot results, larger registrational trials or expansion cohorts in even earlier stages of plasma cell dyscrasias may be initiated. Investigators are looking at the 7 NINT cases to determine if specific biomarkers (like absolute lymphocyte count expansion) can predict and prevent these neurologic events.
Roche / Enspryng (anti-IL6 mAb) / Phase 3 (MOGAD)
The recent data from the Phase 3 METEOROID trial for Enspryng represents a significant milestone in the treatment of Myelin Oligodendrocyte Glycoprotein Antibody-associated Disease (MOGAD). Presented at the 2026 American Academy of Neurology (AAN) Annual Meeting. If approved for the treatment of MOGAD, Enspryng would represent the first authorized treatment for this rare autoimmune condition.
Indication: Myelin Oligodendrocyte Glycoprotein Antibody-associated Disease (MOGAD) is an inflammatory demyelinating condition of the central nervous system (CNS) characterized by the presence of serum antibodies (IgG) that target the MOG protein. Pathophysiologically, MOG is located on the outermost surface of the myelin sheath and oligodendrocyte membranes, making it highly accessible to circulating antibodies. When these autoantibodies bind to their target, they trigger a cascade of complement-mediated damage and cellular infiltration—predominantly by T-cells and macrophages—leading to myelin destruction. Unlike Multiple Sclerosis (MS), MOGAD is pathologically distinct, often showing floppy or large tumefactive lesions and a high predilection for the optic nerves and spinal cord, frequently sparing the brain in adult presentations. The standard of care for MOGAD focuses on the management of acute attacks followed by a risk-stratified approach to maintenance. Acute relapses are treated aggressively with high-dose intravenous methylprednisolone, with early transition to plasma exchange (PLEX) or intravenous immunoglobulin (IVIG) if a full clinical recovery is not achieved. Long-term management is nuanced; because some patients experience a monophasic course (a single lifetime event), maintenance immunosuppression is typically reserved for those with a relapsing phenotype or persistent MOG-IgG seropositivity. For these patients, off-label use of rituximab, mycophenolate mofetil, or monthly IVIG has been the historical mainstay, though the landscape is currently shifting toward targeted biologics, such as IL-6 receptor antagonists, following recent successful clinical trials.
Mechanism: Enspryng is a humanized monoclonal antibody that targets the Interleukin-6 receptor (IL-6R). IL-6 is a key pro-inflammatory cytokine that drives the differentiation of B-cells into antibody-producing plasmablasts and promotes the survival of pathogenic T-cells. By blocking IL-6 signaling, Enspryng suppresses the inflammatory cascade, helps maintain blood-brain barrier integrity, and reduces the production of MOG antibodies that lead to optic neuritis, transverse myelitis, and acute disseminated encephalomyelitis (ADEM).
Trial design: The Phase 3 METEOROID trial was a randomized, double-blind, placebo-controlled study involving adults and adolescents (≥12 years old) with MOGAD who had experienced at least one relapse in the prior year or two in the prior two years. Participants received either satralizumab (120 mg subcutaneous injection) or placebo, with or without background immunosuppressive therapy.
Data: The trial met its primary and key secondary endpoints with high statistical significance. At 48 weeks, 87% of the Enspryng group remained relapse-free compared to 67% in the placebo group (p=0.0025). Enspryng reduced the risk of a new relapse by 68% compared to placebo and demonstrated a 66% reduction in ARR (p=0.0030). There was a 79% reduction in the annualized rate of active MRI lesions and a 73% lower proportion of patients requiring rescue therapies (e.g., steroids or plasma exchange). The safety profile was consistent with Enspryng’s established record in NMOSD. Common adverse events (>5%) included injection-related reactions, influenza, and arthralgia.
Impact:
First-in-Class Potential: There are currently no FDA-approved treatments for MOGAD; clinicians often rely on off-label immunosuppressants like rituximab or mycophenolate mofetil.
Rapid Onset: The treatment effect was observed as early as 8 weeks, providing rapid stabilization for high-risk patients.
Disability Prevention: Since disability in MOGAD is primarily driven by acute relapses rather than gradual progression, the high relapse-free rate directly correlates to better long-term functional outcomes and less reliance on high-dose “rescue” corticosteroids.
Next steps: The trial sponsor has announced plans to submit the METEOROID data to global regulatory authorities (FDA, EMA) in 2026. The drug already holds Orphan Drug Designation, which may expedite the review process. Continued monitoring of the open-label extension phase will be crucial to assess long-term safety and the durability of the 87% relapse-free rate beyond the one-year mark.
Descriptive data releases without numerical data
AstraZeneca / Ultomiris (anti-C5 mAb) / Phase 3 (IgAN): The interim analysis met its first co-primary endpoint with “statistically significant and clinically meaningful” reduction in UPCR was observed as early as Week 10. The Phase 3 LUNA trial met its primary endpoint for proteinuria reduction at the 34-week mark (specific percentage data to be presented at a forthcoming medical meeting). The profile was consistent with Ultomiris’s established record in PNH and aHUS. No new safety signals were identified, though the drug maintains its Boxed Warning regarding the risk of serious meningococcal infections. The trial sponsor confirmed they will seek accelerated regulatory approval in key global markets (US, EU, Japan) based on this Week 34 proteinuria data. The trial will continue to its 106-week completion to evaluate the second co-primary endpoint: the long-term stabilization of kidney function (eGFR). Detailed results are expected to be presented at an upcoming major nephrology conference (likely ERA-EDTA or ASN Kidney Week).
AstraZeneca / Tozorakimab (anti-IL33 mAb) / Phase 3 (COPD): The recent clinical successes for tozorakimab (presented in March and April 2026) represent a major milestone in respiratory medicine. This is the first IL-33-targeting biologic to demonstrate success in Phase 3 trials for COPD, notably succeeding where other IL-33 and ST2 inhibitors had previously failed. The trial sponsor reported that the trials were “statistically significant and highly clinically meaningful.” Crucially, tozorakimab showed benefit regardless of a patient’s blood eosinophil count. This distinguishes it from Dupixent (dupilumab), which is primarily effective in the eosinophilic (Type 2) subgroup. The drug was generally well-tolerated. The safety profile across all three trials was consistent with earlier Phase 2 data, with no major unexpected safety signals reported. Currently, biologics like Dupixent only address about 20–30% of the COPD population (those with high eosinophils). Tozorakimab has the potential to treat up to 80% of the total COPD population, including those without Type 2 inflammation. By targeting the oxidized form of IL-33, tozorakimab is the first therapy to directly address the mucus hypersecretion that leads to the “smoker’s cough” and obstructive symptoms. The trial sponsor plans to share the full data with global regulators (FDA, EMA) in 2026 for potential approval. The PROSPERO trial, a 104-week long-term extension study, is ongoing, with additional results expected in H1 2026. Beyond COPD, tozorakimab is in Phase 3 for severe viral lower respiratory tract disease and Phase 2 for asthma.
To contact us, please send us an email at biotechreadout@gmail.com
Disclaimers
Investigational Status Disclaimer
The therapeutic candidates discussed in this newsletter are currently in clinical development and have not been approved for commercial sale by the U.S. Food and Drug Administration (FDA), the European Medicines Agency (EMA), or other global regulatory authorities. Their safety and efficacy have not been established. References to pipeline products and ongoing clinical trials involve significant risks and uncertainties. Statements regarding the potential safety, potency, or efficacy of investigational drugs reflect current hypotheses and are not a guarantee of future performance or regulatory clearance. The outcome of clinical trials is inherently unpredictable, and clinical results from earlier stages may not be predictive of results in later, larger-scale trials.
No Medical Advice Disclaimer
This newsletter is for informational and educational purposes only. The content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or another qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read in this publication.
No Patient-Provider Relationship Disclaimer
The information provided in this newsletter is for educational and analytical purposes only. Receipt of this information, or any interaction with this content, does not create a physician-patient, pharmacist-patient, or any other professional-provider relationship between you and the authors or publishers. This newsletter should not be used as a substitute for a personal consultation with a qualified healthcare professional.
Forward-Looking Statements Disclaimer
This newsletter contains “forward-looking statements” regarding future events, including clinical trial timing, regulatory milestones, and projected market performance. These statements are based on current expectations and assumptions that are subject to significant risks and uncertainties. Actual results may differ materially from those expressed or implied. We undertake no obligation to update these statements as a result of new information or future developments.
Third-Party Links & Content Disclaimer
This newsletter contains links to third-party websites, including clinical trial registries and corporate presentations. Biotech Readout does not endorse, guarantee, or assume responsibility for the accuracy or reliability of any information offered by third-party providers.
Errors and Omissions Disclaimer
While we strive for technical accuracy, the information in this newsletter is provided on an “as is” basis with no guarantees of completeness, accuracy, or timeliness. Biotech Readout assumes no liability for any errors or omissions in the content of this publication.
Non-Endorsement Disclaimer
Any reference to specific commercial products, processes, or services by trade name, trademark, or manufacturer does not constitute or imply an endorsement or recommendation by the author. All trademarks are the property of their respective owners.
No Investment Advice Disclaimer
This newsletter is for informational purposes only and does not constitute financial, investment, or legal advice. The author is not a registered investment advisor. You should consult with a professional financial advisor before making any investment decisions. The biotechnology sector is highly volatile; past performance is not indicative of future results.
Conflict of Interest Disclaimer
The author of this newsletter maintains a position of independence. At the time of publication, the author holds no direct financial interest, equity, or options in any of the companies mentioned in this report. No compensation has been received from any third party to feature or analyze specific therapeutic candidates or corporate entities.