
Obesity, Part 2
Evolution of treatments: from dark age to golden age

Disclaimer: This newsletter is for educational and informational purposes only and does not constitute medical, investment, or financial advice, nor does it establish a provider-patient relationship. Content may include forward-looking statements and discussions of investigational therapeutic candidates that are not FDA/EMA approved; their safety and efficacy remain unestablished and clinical outcomes are unpredictable. While we strive for accuracy, all information is provided as is without guarantees. This newsletter is independent, and the author holds no financial positions in the companies mentioned nor receives third-party compensation for this coverage. Please find a complete version of our disclaimers linked here.

If you are a paid subscriber, go to this article’s premium readouts here ⬇️
If you are a free reader or subscriber, enjoy the whole article free of charge and consider upgrading for premium charts & analysis on the obesity landscape.
Introduction
In Part 1 of this series on obesity, we traced the evolution of obesity from a perceived moral failing to a recognized chronic disease, explored the hormones like leptin and ghrelin that govern our weight, and shared the genetic breakthroughs that are finally rewriting the willpower myth. In Part 2, we explore the evolution of obesity treatments and how the field climbed out of a century-long dark age and into its current golden age.
The Dark Ages of Obesity Medicines
The first described attempts at producing weight loss are those of Soranus of Ephesus, a Greek physician, in the second century AD. He believed that health depended on the tension of the body’s internal pores. Disease occurred if pores were too tight (strictum) or too loose (laxum). Obesity was viewed as a state of congestion and sluggishness. To fix it, the physician had to loosen and evacuate the system to restore flow. He prescribed elixirs of laxatives and purgatives, as well as heat, massage, and exercise. This remained the mainstay of treatment for well over a thousand years.
If you’re wondering why this specific protocol lasted over a millennium, the credit (or blame) goes to Caelius Aurelianus. In the 5th century, Aurelianus translated Soranus’s Greek works into Latin. His text, On Chronic Diseases, became the standard medical textbook for the Middle Ages. Because the Roman Empire’s medical infrastructure collapsed, the Church and later medieval scholars clung to these Latin translations as “absolute truth.” While the “pore” theory eventually died out, the core components Soranus identified (metabolic stimulation, physical exertion, and caloric restriction) formed the foundations of every wellness movement from the Renaissance until the discovery of the calorie in the 19th century.
Yet, these interventions appear quaint in contrast to the three-headed hydra of horrifying and now banned obesity medications that plagued the 20th Century:
Thyroid Tablets (1920s–1938); banned as a weight loss treatment in 1938 due to potentially fatal cardiac and CNS/PNS toxicity.
Uncoupling Agents (1933-1938) like dinitrophenol (DNP); banned as a weight loss treatment in 1938 due to drug-related cataracts and potentially fatal hyperthermia. DNP was, in fact, a volatile industrial chemical used in French munitions factories to sensitize artillery shells; its weight-loss potential was only discovered when factory workers began to unexpectedly lose weight.
Amphetamines/“Rainbow Pills” (1930s-1997), banned as a weight loss treatment in late 1960s and 1997 due to cardiac and neuropsychiatric toxicity.
I won’t cover their history, and will simply note that they are all banned for very good reasons; they led to the death and disability of thousands and stopping them required the expansion of the FDA’s legal mandate. The fallout from these medical misfires harmed patients and created a safety trauma that dictated FDA policy and clinical skepticism for over half a century. In retrospect, it’s no wonder than it took until 2013 for the American Medical Association (AMA) to formally recognize obesity as a disease.
Since the banned drug classes listed above were often marketed to people seeking a slimmer figure rather than those with clinical illness, the medical community began to view obesity treatment as an elective, cosmetic pursuit. This led to a higher bar for safety; if a drug is for a lifestyle choice, even a 1% risk of a serious side effect (like blindness or heart failure) was considered unacceptable. After the high profile withdrawal of Fen-Phen in 1997, the FDA’s advisory committees became famously risk-averse. For nearly two decades, they rejected or delayed review of now approved medicines like Qsymia or Contrave if they showed even minor cardiovascular signals, fearing a repeat of the “Rainbow Pill” deaths. The perceived danger of pills drove the medical community toward Bariatric Surgery. For a long time, cutting into a healthy organ was seen as safer and more medically rigorous than prescribing a pill, largely because of the obesity’s checkered past of pharmaceutical toxicity.
A Surgical Approach to Obesity
The history of bariatric surgery is a story of serendipitous discovery, evolving from extreme malabsorptive procedures to modern metabolic interventions. It was born out of the observation that patients who underwent intestinal resections for cancer or trauma often suffered from significant, unintended weight loss.
The first intentional surgeries for obesity were designed to prevent the body from absorbing calories by bypassing large sections of the small intestine. In 1954, Dr. Viktor Henrikson performed the first surgery specifically designed to treat obesity in Gothenburg (Göteborg), Sweden. Henrikson’s operation in Gothenburg was an extensive small bowel resection (literally removing about 105 cm of the intestine) rather than a bypass. He theorized that physically shortening the gut would limit caloric absorption. The related Jejunoileal Bypass (JIB) procedure, where the intestine is rerouted rather than removed, was developed shortly after in the United States. Dr. Richard Varco performed the first JIB in 1953 at the University of Minnesota, followed by a famous published report by Kremen, Linner, and Nelson in 1954 at Mount Sinai Hospital in Minneapolis. While patients lost massive amounts of weight, the “blind loop” of bypassed intestine caused severe complications, including chronic diarrhea, electrolyte imbalances, and bypass enteritis (liver failure). By the late 1970s, the JIB was largely abandoned due to its high morbidity.
The surgical field turned away from the intestines and towards the stomach. In 1966, Dr. Edward Mason (often called the “Father of Bariatric Surgery”) at the University of Iowa noticed that women who had a partial gastrectomy for peptic ulcers remained thin. Mason developed a procedure that combined restriction (making the stomach smaller) with malabsorption (bypassing a portion of the small intestine), known as the Roux-en-Y Gastric Bypass (RYGB). The RYGB proved to be far safer than the JIB. It reduced the stomach to a small pouch, limiting how much a person could eat, while the bypass altered hunger hormones.
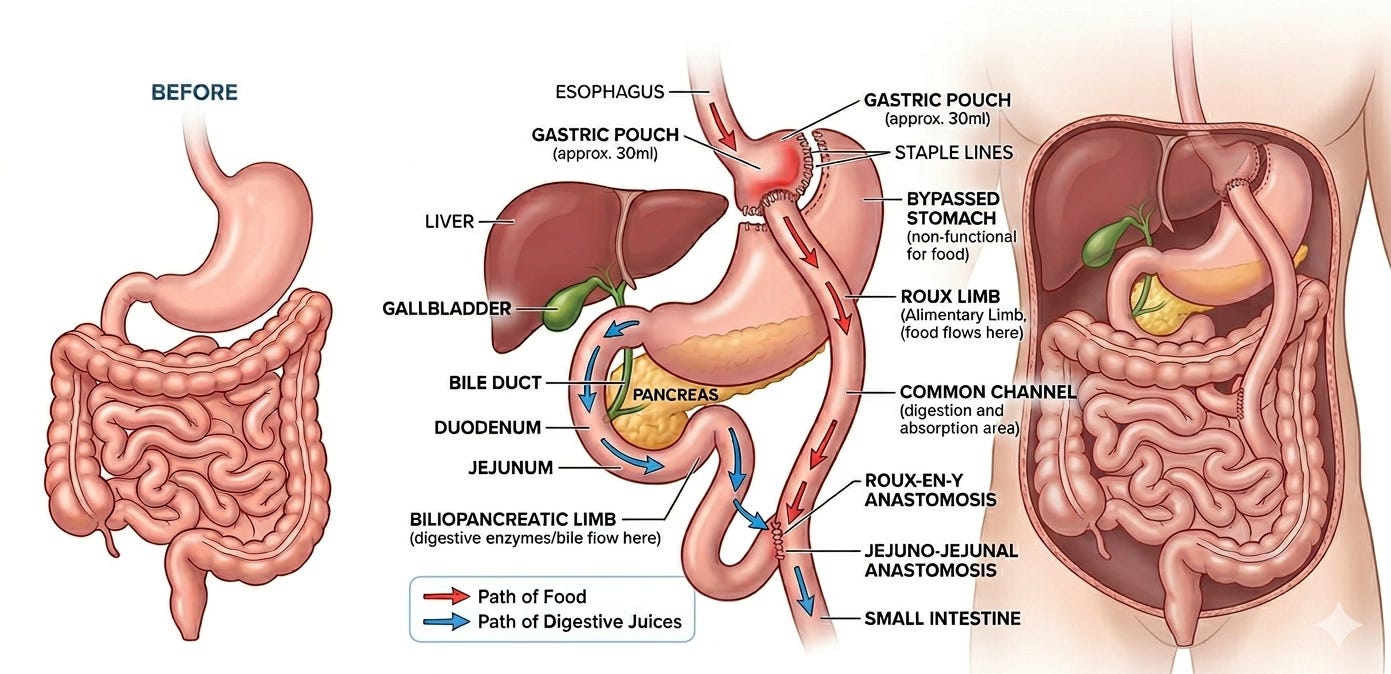
In the 1980s–1990s, surgeons began looking for ways to lose weight without rerouting the intestines, focusing solely on shrinking the stomach. The Vertical Banded Gastroplasty (VBG), also developed by Dr. Mason, involved stapling the stomach to create a small pouch and using a plastic band to slow the exit of food. It became popular but often failed long-term as patients circumvented the surgery with high-calorie liquids. Adjustable Gastric Banding (“Lap-Band”) was introduced in the 1990s. This was an inflatable silicone ring placed around the top of the stomach. It was popular because it was reversible and didn’t involve cutting the stomach, but it fell out of favor due to high rates of “band slippage” and inadequate long-term weight loss.
A big turning point in the history of bariatric surgery was the shift from open surgery (large incisions) to laparoscopy (keyhole surgery). In 1994, the first laparoscopic gastric bypass was performed. This reduced recovery time from weeks to days and significantly lowered the risk of infections and hernias, making the surgery accessible to a much wider population.
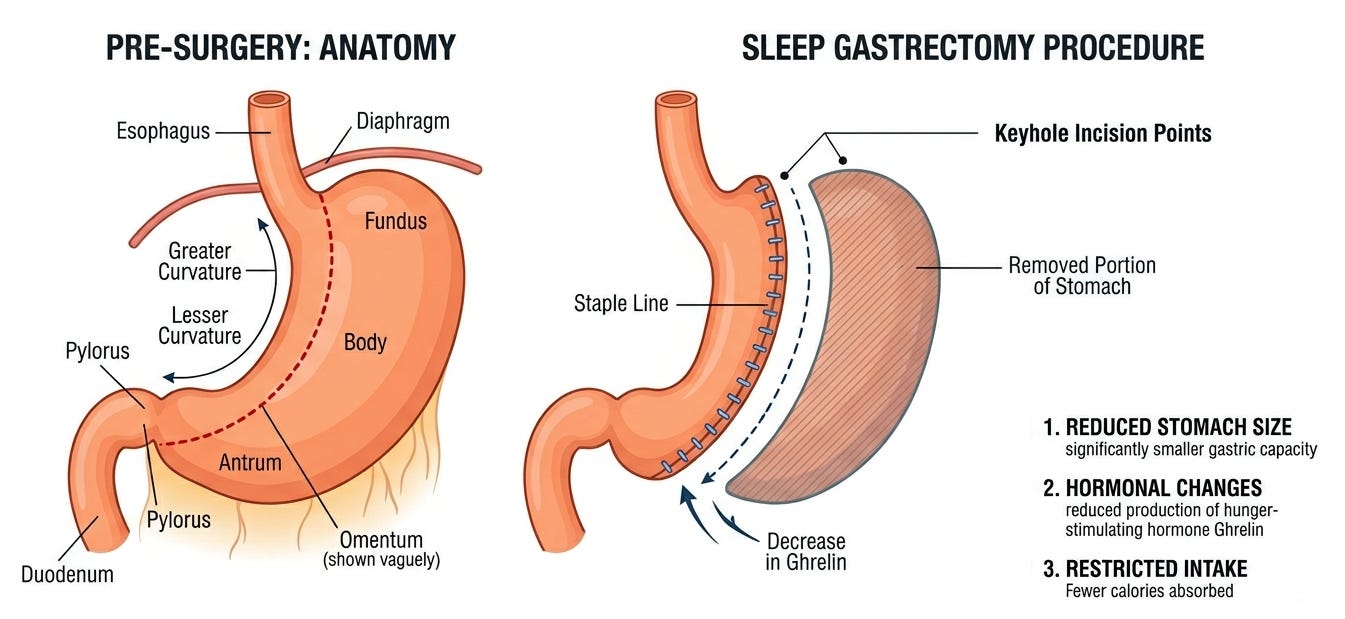
Today, the Sleeve Gastrectomy is the most common bariatric procedure performed worldwide. It is simpler than the bypass and has a lower risk of long-term vitamin deficiencies. It’s also a classic example of a serendipitous medical breakthrough. It wasn’t originally designed as a standalone surgery. Rather, it was a safety measure for high-risk patients that worked far better than anyone expected.
To understand the Sleeve Gastrectomy, you have to look at its predecessor, the Biliopancreatic Diversion with Duodenal Switch (BPD-DS). Developed in the late 1980s by Dr. Douglas Hess, this was a massive, high-risk operation that involved both a sleeve stomach and a significant intestinal bypass. While highly effective for weight loss, it was physically taxing and took many hours to perform. For patients with a BMI over 60 or those with severe heart disease, the long anesthesia time and complexity were often lethal.
In the late 1990s and early 2000s, surgeons including Dr. Michel Gagner at Mt. Sinai in New York began looking for a way to make the BPD-DS safer. They came up with a two-stage approach:
Stage 1: Perform just the stomach reduction (the “Sleeve”) to induce initial weight loss and make the patient healthier.
Stage 2: After 12–18 months, once the patient had lost 50–100 lbs, bring them back to perform the complex intestinal bypass part of the surgery.
The “Aha! moment” occurred when patients started failing to show up for their second-stage appointments. Surgeons realized that many of these Stage 1 patients were losing massive amounts of weight, and keeping it off, without ever having the intestinal bypass. It was originally thought the Sleeve worked solely through restriction (a smaller stomach). However, researchers discovered that removing the fundus (the upper curve of the stomach) also removed the primary production site of ghrelin, the hunger hormone. This meant the Sleeve Gastrectomy wasn’t merely a mechanical tool, but a neuroendocrine intervention.
By the mid-2000s, the data was undeniable. In 2006, the first International Consensus Summit on Sleeve Gastrectomy was held, and by 2010, major surgical societies officially recognized it as a primary, standalone procedure. By 2026, the Sleeve Gastrectomy accounted for over 60% of all bariatric surgeries globally. Its rise was driven by:
Simplicity: It does not involve rerouting the intestines or placing foreign objects (like the Lap-Band).
Pylorus Preservation: By keeping the stomach’s exit valve (the pylorus) intact, it avoids “Dumping Syndrome,” a common and unpleasant side effect of the Gastric Bypass.
Safety Profile: It carries a significantly lower risk of the internal hernias and long-term vitamin deficiencies that plagued the JIB and BPD eras.
The discovery of the Sleeve Gastrectomy shifted the entire surgical philosophy from “how much can we bypass?” to “how can we best manipulate hunger hormones?”
The Golden Age of Obesity Medicines
The contrast between the 20th-century Dark Ages of Obesity Medicines and the current Golden Age of GLP-1/GIP receptor agonists represents a shift from systemic toxicity to molecular precision, as well as a return to hormone biology. While early interventions tried to brute force weight loss by overstimulating the heart or overheating the body, modern assets mimic the body’s natural signaling pathways to regulate appetite and metabolism.
Thyroid tablets and mitochondrial uncouplers worked by creating a state of artificial disease (hyperthyroidism or uncoupled cellular respiration). They forced the body to burn energy by damaging its efficiency. Amphetamines worked by triggering a fight-or-flight stress response to suppress hunger. In contrast, current Golden Age is defined by the FDA-approved incretin mimetics semaglutide (marketed as Wegovy for obesity and Ozempic for Type 2 diabetes) and tirzepatide (marketed as Zepbound for obesity and Mounjaro for Type 2 diabetes). They mimic hormones naturally secreted by the gut after a meal to promote satiety (GLP-1 and GIP). Instead of stressing the system, they signal the brain’s satiety centers (hypothalamus) to feel full and slow gastric emptying, working with the body’s regulatory hardware rather than against it.
As of 2026, the medical community has transitioned from cautious observation to broad, enthusiastic adoption of semaglutide and tirzepatide. These drugs are no longer viewed as short-term fixes but as foundational therapies for Adiposity-Based Chronic Disease (ABCD). Major medical organizations have officially shifted their treatment algorithms to prioritize these assets:
American Diabetes Association (ADA) 2026 Standards: The ADA now recommends semaglutide and tirzepatide not just for glucose control, but specifically for comorbidity management. They are now first-line recommendations for patients with diabetes who also have obesity, heart failure (specifically HFpEF), or chronic kidney disease.
American Association of Clinical Endocrinology (AACE) 2025/2026 Update: The AACE has reframed obesity as a “neuroendocrine disease.” Their latest guidelines prioritize “second-generation” anti-obesity medications (semaglutide/tirzepatide) over older stimulants due to their superior weight loss (20-22% average) and metabolic “reset” capabilities.
NICE (UK) & Global Access: By April 2026, the UK’s NHS has begun a massive multi-year rollout of tirzepatide, acknowledging its cost-effectiveness in preventing long-term complications like stroke and heart attack.
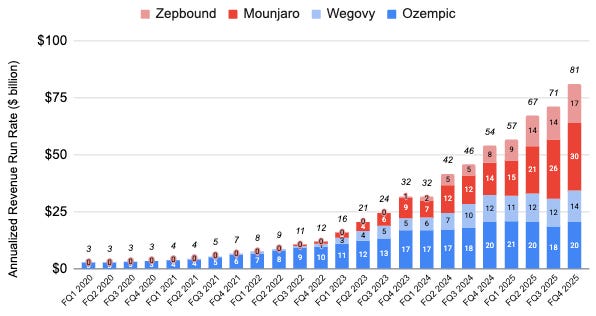
As of April 2026, the combined annualized revenue run rate of semaglutide (marketed as Wegovy for obesity and Ozempic for Type 2 diabetes) and tirzepatide (marketed as Zepbound for obesity and Mounjaro for Type 2 diabetes) exceed $80 billion per year.
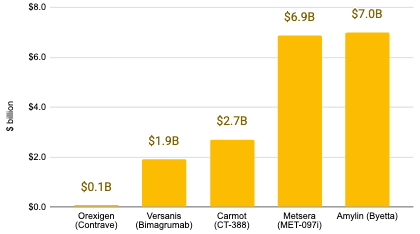
The obesity space has also seen a number of multi-billion dollar acquisitions, consisting mostly (but not exclusively) of incretin drugs.
The discovery of incretins, a family of hormones that GLP-1 and GIP belong to, took over a century. It involves a slow and steady progression from vague physiological observations to the precise molecular engineering of drugs like Wegovy and Zepbound.
Sugar Rush
In Part 1, we discussed two hormones that are linked to body weight and counterbalance each other: ghrelin (hunger signal) and leptin (satiety signal). However, they aren’t the only metabolic hormones on the block. We can’t discuss incretins before first taking a detour into the sugar handling hormones: insulin (sugar storage) and glucagon (sugar moblization). They are the breadcrumbs that ultimately led researchers to the discovery of incretins.
The story starts in the 1920s with Frederick Banting and Charles Best beginning their work at the University of Toronto. They were investigating treatments for Type 1 Diabetes, a disease in which the body fails to produce insulin a persistent autoimmune response kills the insulin-producing beta cells (different from Type 2 Diabetes where patients produce insulin but have become insensitive to it). At the time, Type 1 Diabetes was a death sentence. Banting and Best set out to isolate an internal secretion from the pancreas that could regulate blood sugar, a feat that had frustrated scientists for decades. Before Frederick Banting started, others had tried to isolate insulin from pancreatic extracts but would fail because the pancreas is a dual-purpose organ: it contained both insulin and digestive enzymes, which would chew up the insulin in crude extracts. To solve this, they tied off the pancreatic ducts of living dogs so that the acinar cells died (which produce digestive enzymes), then created extracts from the resulting tissue. To prove it worked, they had a second group of dogs whose entire pancreata had been removed, making them severely diabetic. When these dogs were near death from high blood sugar, Banting and Best injected their extract. Within hours, the dogs’ blood sugar plummeted, and their strength returned. One dog, famously known as Dog 92 (or Marjorie), was kept alive for 70 days using these extracts, a record at the time. They found insulin, but what about glucagon?
The discovery of glucagon occurred in the exact same experiment, but Banting and Best dismissed it as nothing more than a pesky impurity. They noticed that immediately after injecting the pancreatic extract, there was often a brief, sharp increase in blood glucose before the insulin took over and lowered it. In 1923, Charles Kimball and John Murlin at the University of Rochester realized this wasn’t a mistake or a side effect of the insulin. They isolated this “hyperglycemic factor” and named it glucagon, derived from “glucose agonist.” For the next 30 years, much of the scientific community remained skeptical. Many believed glucagon was just damaged insulin or a byproduct of the extraction process rather than a hormone in its own right. In 1953, glucagon believers were vindicated when researchers at Eli Lilly (led by Alfred Staub) successfully crystallized glucagon in its pure form, proving once and for all that it was a distinct protein.
In the 1970s, Dr. Roger Unger at the University of Texas Southwestern Medical Center proposed a revolutionary idea: diabetes isn’t just an insulin deficiency; it’s a bihormonal disease. His central argument was that high blood sugar is not caused by a lack of insulin alone, but by the combined effect of insulin deficiency and glucagon excess. Dr Unger showed that excess glucagon was present in both Type 1 and Type 2 diabetes patients. Despite the strong scientific logic behind Dr. Unger’s bihormonal hypothesis, pure Glucagon Receptor Antagonists (GRAs) are not currently used in standard clinical practice to treat diabetes. Several candidates reached Phase 2 and Phase 3 clinical trials over the last decade, but these programs faced recurring safety hurdles that have prevented FDA approval (liver enzyme elevation, LDL cholesterol increase, alpha-cell hyperplasia, dangerous blood sugar spike on withdrawal).
A Fishing Expedition
While Dr. Unger’s bihormonal hypothesis didn’t result in a major therapeutic win, it did elevate the profile of glucagon, leaving breadcrumbs for other researchers to follow. One of those researchers was Dr. Joel Habener, an endocrinologist at Massachusetts General Hospital. Like Dr. Unger, Dr. Habener believed that if he could understand how glucagon was made, scientists might be able to develop a way to block it, thereby helping diabetics keep their blood sugar from spiking.
By the 1970s, it was becoming clear that many small hormones don’t start out that way. They are originally part of a much larger precursor protein that gets snipped into smaller, active pieces by enzymes. Habener wanted to find the proglucagon gene to see exactly how the body cuts the protein to create glucagon. He suspected that by mapping the gene, he might find other hidden peptides that also played a role in metabolism and could treat diabetes.
Since of the NIH moratorium on cloning mammalian DNA, Dr. Habener turned to the cold-blooded anglerfish (Lophius americanus), a creature that offered a unique anatomical advantage for a molecular biologist. Anglerfish has uniquely large clusters of endocrine tissue called Brockmann bodies. These organs are essentially glucagon factories, making it much easier to isolate the mRNA he needed compared to traditional lab animals. Dr. Habener’s team (including researchers like Graeme Bell and Lund) created a cDNA library from the Brockmann bodies and, since the partial amino acid sequence of glucagon was already known, they fashioned an radioactive probe to find colonies of transformed bacteria that contained the proglucagon gene. Once the correct cDNA was isolated, they used the Sanger sequencing method to determine its genetic code.
They expected a code long enough for the 29 amino acids of glucagon plus a small leader sequence, but instead they found a a massive open reading frame. By looking identifying coding regions for Lysine-Arginine pairs, Dr. Habener and his team could see exactly where cellular enzymes (prohormone convertases) were supposed to snip the long protein chain to release the individual hormones. To their surprise, the gene didn’t only code for glucagon. It also contained sequences for two additional, previously unknown peptides. These were named glucagon-like peptide 1 (GLP-1) and glucagon-like peptide 1 (GLP-2).
A Cut Above the Rest
The transition from identifying a gene sequence to finding a functional medicine was the most contentious period in GLP-1 history. After Dr. Joel Habener cloned the proglucagon gene, the scientific community was stuck: they had the code for GLP-1, but when they synthesized the full peptide (the 1–37 amino acid string), it did absolutely nothing. The breakthrough in the mid-1980s required a shift from genetics to protein chemistry to identify how the body actually cuts the protein to make it active.
Svetlana Mojsov, a chemist at Massachusetts General Hospital, played the pivotal role here. She noticed that the full 37-amino-acid sequence of GLP-1 contained internal dibasic sites, specific amino acid pairs Lysine and Arginine, that act as molecular “cut here” signs for cellular enzymes (prohormone convertases). Mojsov predicted that the first 6 amino acids were essentially a “spacer” and that the body clipped them off to create a shorter, active version: GLP-1 (7–37). She manually synthesized this truncated version, which was a massive technical feat at the time. Working with Habener and a young researcher named Daniel Drucker, they tested this shortened version on rat insulinoma cells. Unlike the full-length version, the 7–37 version triggered a massive release of insulin.
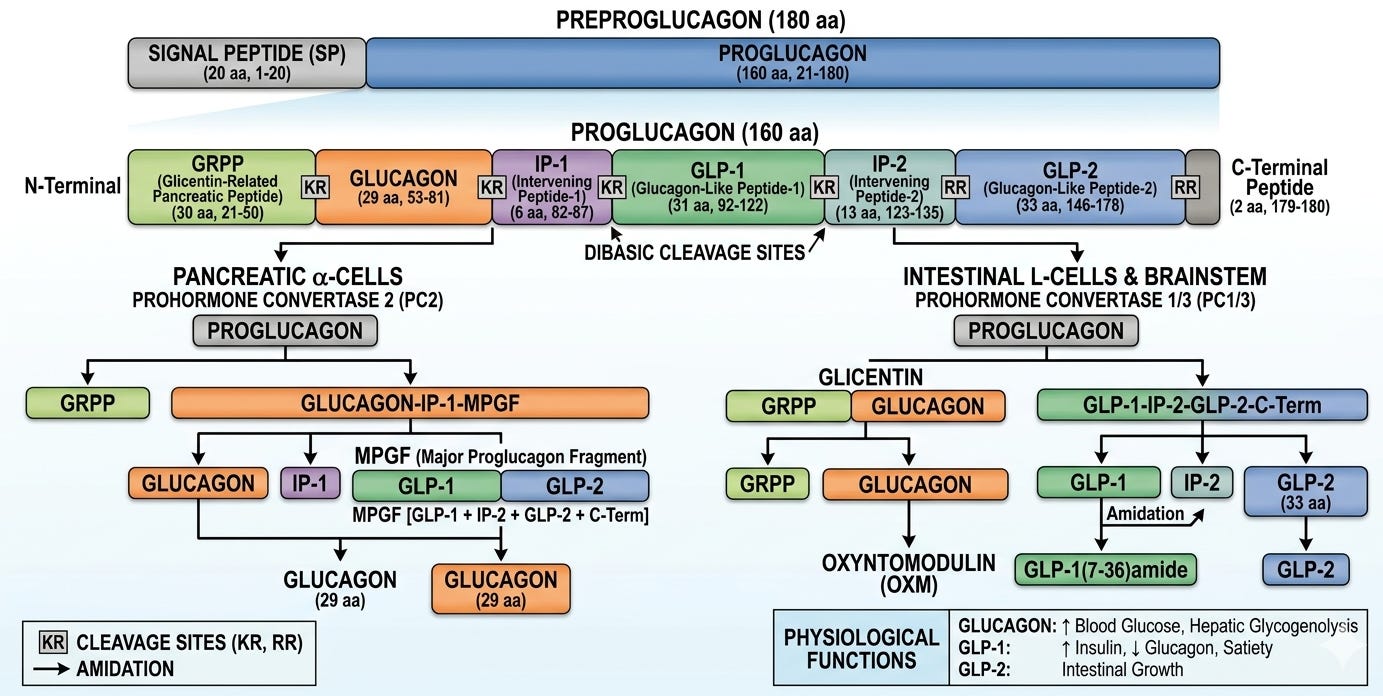
While the Boston team was looking at the gene, Dr. Jens Juul Holst in Copenhagen was coming from the clinical side. He was fascinated by the “Incretin Effect”, the fact that sugar consumed orally triggers a much larger insulin response than sugar injected directly into the blood. Dr. Holst was studying “dumping syndrome” patients (who had parts of their stomachs removed) and found a substance in their gut that stimulated insulin. He independently isolated the truncated GLP-1 from human intestinal tissue. His work proved that GLP-1 was a real hormone produced by the L-cells of the human gut in response to food.
Research from these two groups culminated in a series of landmark papers in 1987. The PNAS paper, led by Daniel Drucker, Svetlana Mojsov, and Joel Habener, was the first to show that GLP-1 (7–37) stimulates insulin gene expression and increases cAMP levels in pancreatic cells, while the full-length version (1-37) was inert. Months later, Dr. Jens Juul Holst published in FEBS Letters, confirming that the gut actually secretes this specific 7-37 version into the bloodstream after we eat and identifying it as a natural human incretin. These researchers jointly received the Lasker Award in 2024 for their contributions to GLP-1 based therapy for obesity.
Most importantly from a therapeutic standpoint, these studies showed that GLP-1 (7–37) was the most potent insulin-stimulating hormone ever discovered and that it only stimulated insulin when blood sugar was high. This was considered a “Holy Grail” of diabetes research. Unlike the older diabetes drugs (sulfonylureas) that forced insulin out and caused dangerous crashes (hypoglycemia), GLP-1 was smart; it stopped working as soon as blood sugar returned to normal.
Unfortunately, there was a major roadblock that accounted for the almost 20-year delay between the 1987 discovery of GLP-1 to the first FDA-approved drug in 2005: its short half-life (roughly 1.5 to 2 minutes). In its natural state, human GLP-1 only lasts a few minutes in the bloodstream. This extreme instability is not a biological mistake, it is a precision-engineered safety feature of the body to prevent life-threatening drops in blood sugar (hypoglycemia) once a meal has been processed.
The perpetrator was DPP-4 (Dipeptidyl Peptidase-4), an enzyme known to immunologists for decades as CD26. Its role as the assassin of GLP-1 wasn’t realized until the 1990s. DPP-4 was ubiquitous, sitting on the surface of blood vessels, kidneys, and floating freely in the plasma and waiting for a substrate to pass by. In 1993, Rolf Mentlein and his team at Kiel University in Germany performed a definitive study. They incubated GLP-1 in human serum and observed that the enzyme DPP-4 was specifically snipping off the first two amino acids (Histidine-Alanine) from its front end. When they added a DPP-4 inhibitor to the test tube, the GLP-1 remained intact, suggesting that DPP-4 was indeed responsible for GLP-1 degradation.
Mentlein’s discovery created two competing paths for the biotech industry:
The Inhibitor Path: Create a pill that blocks the enzyme throughout the whole body to let your natural GLP-1 last longer. Nancy Thornberry and the team at Merck led the charge here, developing a DPP-4 inhibitor that could be taken as a once-daily pill. In 2006, Januvia became the first of its class approved by the FDA.
The Bypass Path: Create a version of GLP-1 that the enzyme couldn’t recognize. This path led to the development of long-acting GLP-1 agonists, but it was by no means a straight shot to success.
I hope you have been enjoying this two-part series on obesity. Don’t forget to subscribe for free so you don’t miss out on future posts.
False Start
In 1987, an assistant professor of medicine at Harvard Medical School (HMS) and chief of the Diabetes Unit at Harvard-affiliated Beth Israel Hospital by the name of Dr. Jeffrey S. Flier had a meeting that would set a chain of events in motion that could have changed the course of medicine forever but did not, due to hodgepodge of idiosyncrasies that often befall biotech startups. A fellow endocrinologist, Dr. John Baxter from UCSF, happened across Dr. Flier who pitched him on an idea he had been kicking around: a nasal spray formulation with insulin (no, not GLP-1). Unlike investors who had balked at Dr. Flier’s idea, Dr. Baxter was intrigued an invited Dr. Flier to kickstart a development company in his recently founded biotech startup CalBio.
Dr. Flier wasted no time at all. He called up former colleague Ron Kahn (director of research at the Joslin Diabetes Center) and Bruce Spiegelman (another HMS professor), and proceeded to have “several memorable weekend sessions in [his] living room discussing potential project areas”. In collaboration with Dr. Baxter, the trio decided to spin up Metabolic Biosystems (MetaBio), a wholly owned subsidiary of CalBio that was kickstarted by a $30 million cash infusion from Pfizer over five years in exchange for exclusive rights to any clinical-stage programs. Their programs fell into four categories: insulin analogues, enhancers of insulin action, adipocyte secreted factors, and gut factors regulating metabolism. They had lots of ideas, but didn’t yet have a lead program. That would soon change when, in May 1987, Dr. Flier attended the American Society of Clinical Investigation meeting in Atlantic City.
Scanning a “thick book of abstracts”, Dr. Flier identified a Saturday 8:00 AM talk by Joel Habener’s lab at the Chalfonte-Haddon Hall Hotel (that name sounds familiar). Dr. Habener revealed that a new gastrointestinal peptide, GLP-1, enhanced insulin secretion in a glucose-dependent manner. Recognizing the magnitude of the discovery, Dr. Flier approached Dr. Habener immediately. Within weeks, MetaBio secured exclusive worldwide licenses for the GLP-1 patents from MGH and the Howard Hughes Medical Institute (HHMI).
Between 1988 and 1990, the joint team’s human infusion studies in Type 2 diabetic subjects yielded three critical findings:
Strong Glucose Control: Several days of GLP-1 infusion lowered blood sugar significantly in patients with type 2 diabetes.
A “Smart” Mechanism: Unlike traditional insulin shots, it only boosted insulin when glucose was high, drastically reducing the risk of dangerous “lows.”
Weight Loss Potential: Crucially, the team discovered that GLP-1 slowed gastric emptying and reduced hunger—the exact mechanism driving today’s obesity drug craze.
However, this data would never see the light of day. In the 2024 memoir from which this section is sourced, Dr. Flier remarks, “none of this definitive work was ever presented publicly or published, so its existence, until now, has been known only to those involved.” In the next couple of years, the program would shut down and the ownership share of the founding MetaBio trio would be bought out. There were a few reasons that lead to the untimely demise of what seems to be the first GLP-1 agonist program in biotech history:
Pfizer’s lack of conviction in injectable diabetes drugs: Following a presentation of the first-in-human data to Pfizer collaborators Nancy Hutson (then Senior VP of Global R&D at Pfizer’s Groton site) and Gregory Gardiner (then the architect of Pfizer’s external biotechnology strategy), senior management “concluded that there would never be another injectable therapy for diabetes other than insulin”, although their rationale was never explained to Dr. Flier. They told the MetaBio team to come back to them when they had identified an alternative route of administration other than IV, such as trans-nasal or transcutaneous. This was not a viable path, given the short half-life of GLP-1 and the fact that DPP-4 hadn’t been discovered yet (it was discovered a mere 1-2 years after MetaBio was sunsetted). Pfizer ended the deal a year early, leaving the company unfunded.
Cannibalization by their TopCo’s lead program: As a wholly owned subsidiary, MetaBio had to compete for resources against CalBio’s lead asset, Natrecor (Auriculin), a treatment for heart failure. When Pfizer ended its commitment after four years, CalBio CEO Rich Casey faced a choice: hunt for a new partner or prioritize the “safer” bet of Natrecor. Casey later recalled a “herd mentality” on the street. Lacking a powerful internal champion or an independent capital structure to weather the setback, MetaBio’s GLP-1 program was sacrificed to fund Natrecor. While Natrecor eventually led to CalBio’s (renamed to Scios) acquisition by Johnson and Johnson (J&J) for $2.4 billion, a mere rounding error compared to the +$80 billion GLP-1 empire they let slip away.
After MetaBio collapsed, the patents returned to the institutions and were eventually picked up by Novo Nordisk. It took nearly 20 years for Novo Nordisk to achieve what Pfizer deemed impossible, eventually launching liraglutide (approved for obesity in 2014). Today, Novo Nordisk sees almost $35 billion in annual revenue from this class, a significant missed opportunity as a result of Pfizer’s premature exit. While the CalBio/Scios-J&J acquisition appeared to be a success, it represents a staggering Opportunity Cost. They chose a $2 billion exit today over a potentially +$10 billion market tomorrow (Pfizer acquired Metsera’s GLP-1 pipeline for up to $10 billion in November 2025) because they didn’t trust their own data over the prevailing industry dogma.
Monster Mash
The secret success then quiet collapse of MetaBio might have delayed the development of long-acting GLP-1 agonists, but only for a year or so. In the early 1990s, Dr. John Eng, a physician-researcher at the Bronx VA Medical Center, was studying various animal venoms. He was looking for bioactive peptides that might have medical applications. Dr. Eng focused on the Gila monster (Heloderma suspectum), a venomous lizard native to the Southwestern US. He knew these lizards could survive for months on a single meal while maintaining stable blood sugar. In 1992, Dr. Eng isolated a peptide from the lizard’s salivary secretions that he named Exendin-4. He realized that Exendin-4 was roughly 50% identical to human GLP-1. Crucially, the lizard version replaced the vulnerable Alanine at position 8 with a Glycine. This tiny change made the peptide invisible to the DPP-4 scissors, allowing it to last for hours in the blood instead of minutes.
Surprisingly, the discovery was initially met with silence. The Department of Veterans Affairs formally waived its rights to patent Dr. Eng’s discovery, viewing the maintenance fees as an unjustifiable expense. Dr. Eng, convinced of the peptide’s potential, assumed the patent costs out of his own pocket, a rare instance of a government researcher owning a blockbuster molecule. Eventually, a small San Diego biotech startup called Amylin Pharmaceuticals saw the potential. They licensed the peptide from Dr. Eng and began the arduous process of clinical development.
Amylin lacked the resources to bring a major metabolic drug to market alone, so they partnered with Eli Lilly in 2002. The clinical trials (the AC2993 program) demonstrated that twice-daily injections of the synthetic version of Exendin-4, then named exenatide, significantly lowered HbA1c levels. During the trials, researchers noticed a consistent side effect: patients were losing weight. This was the first clinical proof that GLP-1s could be more than insulin secretagogues. They were also potent satiety signals. On April 28, 2005, the FDA approved Byetta (exenatide) as an adjunctive therapy for Type 2 diabetes. It was the first approved GLP-1 receptor agonist in history, but it wouldn’t be the last.
The Hitchhiker’s Guide to GLP-1s
Novo Nordisk scientists followed a different route to subvert DPP-4. Researchers like Knud Jensen and Lotte Bjerre Knudsen set out to modify human GLP-1 so that it stays in the blood longer without changing its shape so much that the body thinks it’s an invader (like the lizard-derived Byetta). In the mid-1990s, the Novo team made a breakthrough in acylation, the process of attaching a fatty acid chain to a peptide. They attached a 16-carbon fatty acid (palmitic acid) to the GLP-1 molecule. This fatty acid acted like a sticky tail that hitchhiked on albumin in the bloodstream.
To make this work, they had to make two specific tweaks to the human GLP-1 sequence:
Amino Acid Substitution: They swapped one amino acid (Lysine for Arginine at position 34) to ensure the fatty acid tail only attached to one specific spot (position 26).
Fatty Acid Tail Linker: They added the C-16 fatty acid chain via a spacer (glutamic acid).
This created a molecule that was 97% identical to human GLP-1 but had a half-life of 13 hours, allowing for a simple once-daily injection (more convenient than the twice-daily shot Byetta). Liraglutide entered clinical trials under the name NN2211. Novo launched a massive clinical program called LEAD (Liraglutide Effect and Action in Diabetes), which consisted of six Phase 3 trials comparing liraglutide to every major diabetes drug on the market including insulin and Byetta. Liraglutide was not only superior in lowering blood sugar but was much better tolerated by patients. On January 25, 2010, the FDA approved Victoza (liraglutide), the world’s first human-sequence GLP-1 analog. During these trials, the weight loss was so consistent that Novo Nordisk decided to study a higher dose specifically for obesity. This led to the approval of Saxenda in 2014, the exact same molecule as Victoza, just at a 3.0 mg dose.
Two is Company
After the success of the once-daily Victoza, Novo Nordisk’s goal was clear: reach once-weekly dosing. To do this, they needed to make the molecule stickier and even more resistant to degradation. They achieved this in two ways:
Fatty Acid Optimization: Scientists replaced the 16-carbon fatty acid used in Liraglutide with a 18-carbon diacid chain. This significantly increased the molecule’s affinity for Albumin, acting like a much stronger anchor.
Amino Acid Shield: They swapped the Alanine at position 8 for Alpha-aminoisobutyric acid (Aib). This change made the N-terminus, the part that DPP-4 usually attacks, completely unrecognizable to the enzyme.
The resulting drug, called semaglutide, had a half-life of approximately 165 hours (7 days). Starting in 2013, the SUSTAIN program (1 through 10) tested semaglutide against nearly every competitor. SUSTAIN-6 (2016) proved that semaglutide significantly reduced major adverse cardiovascular events (MACE) in T2D patients. Ozempic (semaglutide) was approved for Type 2 Diabetes in 2017. Similar to Victoza/Saxenda, Novo recognized profound weight loss in the SUSTAIN trials and launched the STEP trials, leading to the 2021 approval of Wegovy (higher dose of the same active peptide in Ozempic) specifically for chronic weight management in overweight/obesity. Importantly, the SELECT trial demonstrated that Wegovy reduced the risk of death from cardiovascular causes, nonfatal myocardial infarction, or nonfatal stroke (MACE-3) by 20% (HR 0.8, p<0.001) in adults with established cardiovascular disease and obesity or overweight compared to a placebo control. This provided the first proof that weight loss in non-diabeted obese patients could result in an improvement in cardiovascular outcomes, which physicians and insurance companies tend to take more seriously.
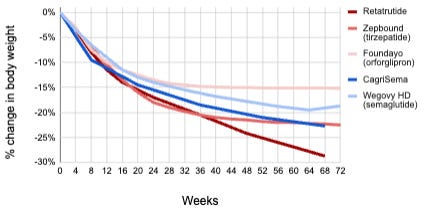
While Novo Nordisk optimized GLP-1, Eli Lilly returned to the bihormonal philosophies of Drs. Roger Unger and Joel Habener. They realized that GLP-1 alone might have a ceiling and decided to pair it with another hormone: GIP (Glucose-dependent Insulinotropic Polypeptide). For years, GIP was considered a forgotten hormone because, unlike GLP-1, it doesn’t work well on its own in diabetics. However, Lilly’s researchers (led by Tamer Coskun) discovered that when you combine GIP and GLP-1, the GIP component seems to prime the adipose (fat) tissue and reduce the nausea often caused by GLP-1. Tirzepatide is a single peptide chain that activates both GLP-1 and GIP receptors. It uses a 20-carbon fatty acid for once-weekly dosing, similar to Novo’s technology but with a more potent weight loss effect in an overweight/obese population (Phase 3b SURMOUNT-5 trial showed -20.2% weight loss in the tirzepatide arm versus -13.7% weight loss in the semaglutide arm after 72 weeks of treatment, p<0.001). Mounjaro was approved for Type 2 diabetes in 2022, and Zepbound for overweight/obesity followed in 2023.
There are a number of other clinical-stage GLP-1/GIP dual agonists including CT-388 by Roche, SYH2082 by AstraZeneca, MariTide by Amgen (actually an anti-GIP antibody fused to a GLP-1 agonist), VK2735 by Viking Therapeutics, PN-458 by Protagonist, and KAI-9531 by Kailera. Others are turning to amylin, another pancreas-derived satiety hormone, in place of GIP for their dual agonists. These include Novo’s CagriSema (GLP-1 and amylin FDC) and Amycretin (dual agonist), and Pfizer’s MET-233i + MET-097i (GLP-1 and amylin FDC).
Three is a Crowd, and Five is a Stampede
Multi-agonists that stack multiple targets on top of GLP-1 are currently making their way through clinical trials. Retatrutide is a single peptide that activates three different receptors: GLP-1, GIP, and Glucagon (GGG). It is the direct successor to the dual-agonist tirzepatide. As we discussed, glucagon was suspected as the enemy in diabetes because it raises blood sugar. However, Eli Lilly’s researchers realized that activating the glucagon receptor in the presence of GLP-1/GIP can increase energy expenditure (calories burned) and specifically reduce liver fat.
In March 2026, Eli Lilly announced positive Phase 3 results from the TRANSCEND-T2D-1 trial. At the 12 mg dose, patients with Type 2 diabetes achieved up to a 2.0% reduction in HbA1c and lost an average of -16.8% of their body weight in just 40 weeks. A prior Phase 2 trial in patients with non-diabetic obesity showed up to -24.2% after 48 weeks of treatment on retatrutide, the most weight loss for any drug to date, as well as normalization in liver fat for in 93% of patients, supporting its ongoing investigation as a potential therapeutic for MASH (Metabolic Dysfunction-Associated Steatohepatitis). Eli Lilly plans to present Phase 3 TRANSCEND data for retatrutide American Diabetes Association (ADA) 2026 meeting from June 5-8, 2026.
Others are developing GGG drugs including BI 3034701 by Boehringer Ingelheim, UBT251 by Novo Nordisk, PN-477 by Protagonist, and KAI-4729 by Kailera. Recently, Eli Lilly has disclosed a quintuple agonist (GLP-1, GIP, Glucagon, Amylin/Calcitonin), with rat data to be presented at the ADA 2026 meeting. As of writing this, a different quintuple agonist (GLP-1, GIP, PPARα/γ/δ) was reported in Nature claiming greater weight loss potency then GLP-1/GIP dual agonism in diabetic induced obese (DIO) mice. Note that results in animal models frequently do not translate to human clinical safety or efficacy, so the jury is out on whether these quintuple agents are competitive, pending data from randomized placebo-controlled clinical trials in human patients with overweight/obesity.
What’s on the Horizon?
While much of the field has been focused on enhancing GLP-1’s weight loss potency, others have sought out complimentary mechanisms that they can stack onto GLP-1 that which add new effects. These include myostatin inhibitors to preserve muscle during weight loss (bimagrumab, emugrobart, taldefgrobep alfa), INHBE knockdown to selectively shed visceral fat (ARO-INHBE, WVE-007, SGB-7342), of FGF21 to enhance the liver fat degradation (MWN105, HEC88473).
The field is also shifting to greater convenience with once-daily oral pills. On December 23, 2025, the Novo Nordisk’s Wegovy pill was approved by the FDA and became the first oral GLP-1 for obesity. It uses a SNAC (salcaprozate sodium) absorption enhancer to protect the semaglutide peptide from being chewed up by stomach acid and enzymes. Nevertheless, the protection that SNAC offers isn’t absolute so the Wegovy pill requires an 8-hour overnight fast with a >30 minute post-dose fast (participants who took the pill after eating had zero or limited measurable exposure to the drug) and it must be taken with no more than 4 ounces (120 mL) of plain, still water (to avoid diluting the SNAC). Approved on April 1, 2026, Eli Lilly’s Foundayo (orforglipron) became the first approved small molecule GLP-1 agonist for obesity, and the only GLP-1 pill for weight loss that can be taken any time of day without food or water restrictions.
Conclusion
The transition of obesity medicine from the Dark Ages of systemic toxicity to the Golden Age of molecular precision is a medical success story. We have moved from a century of trying to brute force weight loss through dangerous stimulants and malabsorptive surgeries to an era of elegant neuroendocrine mimicry.
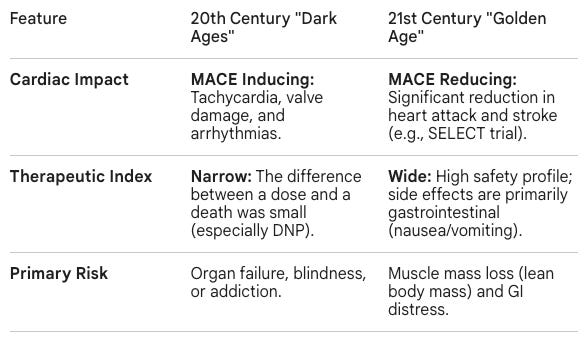
As we move toward the late 2020s, the ceiling for medical weight loss continues to rise. With retatrutide pushing the boundaries of triple-agonism and CagriSema exploring the synergy between the gut and the hindbrain, the distinction between surgical and medical outcomes is blurring. The clinical conversation has shifted from weight loss induction toward the optimization of maximal precision, muscle preservation, organ fat reduction, and quieting the food noise that has long been mischaracterized as a lack of willpower.
However, the Golden Age brings its own set of challenges. As these assets become foundational therapies for a massive global population, the focus of the next decade will likely shift toward durability, accessibility, and personalization. The race is now on to determine which patients benefit most from GIP-heavy versus glucagon-heavy pathways, and how we can maintain these metabolic resets over a lifetime.
Obesity was once a field defined by its failures. Today, it is the vanguard of biotech innovation. By following the breadcrumbs left by the fishing expeditions of the 1980s (literally), we haven’t just found a way to manage weight, we’ve unlocked a deeper understanding of the sophisticated hormonal symphony that governs us all.
Note to readers: I hope you enjoyed this two-part series on obesity as much as I enjoyed writing it. Don’t forget to check out Part 1 if you missed it, and subscribe for free so you don’t miss out on future posts.
Premium Charts & Analysis
Please feel free to email me at biotechreadout@gmail.com with “CHARTS” in the subject line to share suggestions or request chart updates. All charts reflect data current as of the specified date and may not include every drug development program, although we aimed to capture the vast majority.
Press ⬇️ to navigate to a section
Keep reading with a 7-day free trial
Subscribe to Biotech Readout to keep reading this post and get 7 days of free access to the full post archives.