
Obesity, Part 1
Societal perception, recognition as a disease, and the discovery of its hormonal & genetic drivers

Disclaimer: This newsletter is for educational and informational purposes only and does not constitute medical, investment, or financial advice, nor does it establish a provider-patient relationship. Content may include forward-looking statements and discussions of investigational therapeutic candidates that are not FDA/EMA approved; their safety and efficacy remain unestablished and clinical outcomes are unpredictable. While we strive for accuracy, all information is provided as is without guarantees. This newsletter is independent, and the author holds no financial positions in the companies mentioned nor receives third-party compensation for this coverage. Please find a complete version of our disclaimers at the bottom of this article and on our About page.

Introduction
For centuries, the human body has served as a canvas for demonstrating wealth, survival, and social status, shifting from the celebrated curves of the Renaissance to the digitally-perfected thinness of the modern era. But beneath these changing cultural silhouettes lies a complex biological machinery that the world is only just beginning to understand. In Part 1 of our two-part series on obesity, we trace the evolution of obesity from a perceived moral failing to a recognized chronic disease, explore the hormones that govern our weight, and share the genetic breakthroughs that are finally rewriting the willpower myth.
In Part 2, we explore the evolution of obesity treatments and how the field climbed out of a century-long dark age and emerged into the current golden age.
The Weight of Society
The perception of body weight in Western society has undergone a dramatic and complicated transformation, shifting from a celebrated symbol of survival and wealth to a modern marker of health and self-discipline. Historically, this evolution is often summarized as a move from “large as high status” to “thin as high status.”
For most of human history, excess body weight was rare and often seen as a sign of wealth and vitality. Artifacts like the Venus of Willendorf (dated approx. 25,000 BCE) suggest that larger bodies were associated with fertility and the ability to survive food scarcity. In the Renaissance and Baroque periods, artists like Peter Paul Rubens (1577–1640) painted full-figured women as the pinnacle of beauty, leading to the term “Rubinesque”. High body fat signaled that a person was wealthy enough to avoid manual labor and afford a rich diet. In Europe, being portly remained a marker of the elite. However, religious texts occasionally framed gluttony as a moral failing, beginning a long history of social stigma.

{kind=link}
Before the 20th century, most clothes were tailor-made or adjusted at home. The rise of Ready-to-Wear clothing changed the human relationship with size. For the first time, people had to fit into standard sizes rather than clothes being made to fit them. If a garment didn’t fit, the fault shifted from the tailor to the wearer’s body. At the turn of the century, the ideal was still somewhat robust, epitomized by the “Gibson Girl”, a representation of the feminine ideal as depicted in illustrations of artist Charles Dana Gibson. The most distinctive feature of the look was the S-bend corset. Unlike previous corsets that squeezed the waist evenly, the S-bend pushed the chest forward and the hips back. The corset cinched the waist dramatically, creating a sharp contrast between the full bust and the flared hips. The Gibson Girl look would dominate American fashion and social ideals from the 1890s to the early 1910s.

{kind=link}
The 1920s saw one of the most drastic shifts in the history of the Western female silhouette. Following World War I, women dropped the restrictive corset in favor of a straight, tubular look. This silhouette, characterized by a flat chest, dropped waist, and straight lines, was fueled by a desire for liberation after the constraints of WWI. To achieve this, women began binding their chests and dieting aggressively to maintain a youthful frame. The burgeoning film industry began exporting this specific aesthetic globally. On-screen stars like Clara Bow became the visual blueprint for what modernity looked like. The tubular look allowed for freedom of movement. Bow, known for her kinetic screen presence, showed women that fashion didn’t have to be restrictive; you could dance the Charleston and move freely.

{kind=link}
While the 1950s celebrated a curvier ideal (Marilyn Monroe), this was often a managed curviness, a tiny waist achieved through shapewear. Fashion models like Dovima or Suzy Parker were elegant, mature, and possessed womanly “New Look” silhouettes. By the 1960s, the ideal moved from slim to extremely thin. In 1966, the model Lesley Lawson, nicknamed Twiggy, arrived on the scene. Twiggy was only 16 when she became a global sensation. Her frame, which was naturally 5’6” and roughly 91 lbs, represented a radical departure from the hourglass. Her look coincided with the rise of the miniskirt. Leggy, angular thinness became a requirement for the new fashions coming out of London’s Carnaby Street.
The 1970s and 1980s introduced a slightly more athletic version of the thin ideal, but it remained geographically and socioeconomically exclusive. Models like Christie Brinkley and Elle Macpherson (nicknamed “The Body”) promoted a look that was tall, tanned, and fit. While this looked “healthier” than Twiggy, it was actually harder to achieve for the average person. It required a combination of extreme thinness plus muscle tone, a look that required significant time and money spent at the newly popular aerobics studios and gyms. High-fashion designers like Karl Lagerfeld (Chanel) and Gianni Versace began designing clothes that were increasingly narrow, cut specifically for the 5’10”, size 0-2 frame of the Supermodel.
The 1990s saw a reversion to ultra-thin ideals. Kate Moss became the face of this movement. Unlike the 5’11” Amazonian supermodels of the previous decade, she was shorter (5’7”) and possessed a much more delicate look that felt accessible yet elite. The goal was an angular, hanger-like frame. Clothes were often slip dresses or oversized knits that hung loosely off the body, emphasizing thinness by showing how much space was left in the garment. The fashion of this era didn’t exist in a vacuum; it was heavily tied to the grunge music scene emerging from the Pacific Northwest (Nirvana, Pearl Jam). The aesthetic celebrated a lack of grooming. Pale skin, unwashed hair, and dark circles under the eyes were seen as authentic and anti-establishment. In this context, being extremely thin was a way to look like you didn’t care about the mainstream fitness or wellness culture of the 80s. It was a visual rejection of the California tan and the aerobics body.
By the late 1990s, this look became so pervasive that it sparked a massive public health debate. Nutritionists and doctors raised alarms that the “minimalist waif” look was unattainable for most healthy adults and was contributing to a rise in eating disorders. In response to the criticism, the fashion industry slowly began to shift back toward a more athletic “Brazilian” look (led by Gisele Bündchen in the early 2000s), though the baseline for sample size thinness remained much lower than it had been in the 1950s.
The 2000s pushed the thin ideal into the mainstream through the handiwork of Alexander McQueen, then head designer of Givenchy. McQueen first introduced the “Bumster” look in his 1993 Taxi Driver collection and refined it in 1996’s Dante. His goal wasn’t actually about being “sexy” in a traditional sense; he wanted to elongate the spine. By dropping the waistline to the very top of the hip bone, he shifted the visual focus from the waist to the base of the spine. However, when this look trickled down into 2000s mainstream retail resulting in the “Y2K look”: ultra-low-rise jeans and midriff-baring tops that made the stomach and hips the primary area of scrutiny. This created a cultural anxiety around even minor amounts of body fat on the hips and lower abdomenal area, leading to the derogatory term “muffin top.” Professional retouchers used Photoshop to slim down models in magazines. The public knew these images were altered, but they were still the only images available.

{kind=link}
The 2010s marked a dramatic pivot away from the thin ideal. As Instagram (launched in 2010) became the dominant cultural force, the flat silhouette of the previous decades was replaced by a hyper-curvaceous ideal. This decade popularized a very specific silhouette: a tiny waist and flat stomach (the “skinny” legacy) paired with exaggerated hips and a large butt (the “curvy” addition). The middle of the decade saw the rise of “Fitness Inspiration” or “Fitspo.” Thinness was rebranded as strength. Being toned became the new requirement, meaning that not only did one have to be low-weight, but they also had to have visible muscle definition.
The history of weight is a narrative of shifting signals, where the human body has served as a canvas for demonstrating everything from economic prosperity and survival to moral discipline and social status. For centuries, the robust figure was a celebrated symbol of the elite’s abundance, yet the industrialization of the 19th century and the rise of statistical “averages” began a slow march toward the pathologization of body mass. This transformation accelerated in the 20th and 21st centuries, as thinness was rebranded first as a marker of youthful rebellion, then as an indicator of financial wellness, and finally as a digitally-perfected aesthetic that often ignored biological reality. This complex legacy, a mixture of actuarial math, fashion extremes, and socioeconomic signaling, has created a modern paradox: society continues to treat weight as a personal choice and a visual status symbol, even as clinical science increasingly recognizes obesity as a complex, chronic disease driven by metabolic, genetic, and environmental factors rather than simple willpower.
Obesity, Disease or Distraction?
Hippocrates was among the first to recognize obesity as a medical concern, noting that “sudden death is more common in those who are naturally fat than in the lean.” He prescribed exercise and specific diets, though the focus remained largely on “humoral” balance. In the 18th century, conditions like gout (associated with rich foods) were seen as “the disease of kings,” almost a status symbol among the aristocracy. So, how did the medical establishment start to formally recognize obesity as a disease?
It began with the modern medical quantification of weight. Adolphe Quetelet was a 19th-century Belgian mathematician, astronomer, and statistician and is often considered one of the founding fathers of modern sociology. Quetelet was fascinated by the application of probability and statistics to human characteristics. He believed that human traits, both physical and moral, followed a normal distribution (the “Bell Curve”). In the 1830s, Quetelet began analyzing data from French and Scottish soldiers to determine how body weight related to height. He observed several key patterns:
Non-Linear Growth: He realized that weight does not increase in a 1:1 ratio with height (linear). If it did, tall people would be impossibly thin and short people would be impossibly wide.
The Square Law: Through his observations, he determined that after the initial growth spurt of puberty, “the weight increases as the square of the height.”
With these ratios in hand, Quetelet coined the Quetelet Index, or what we now refer to as the Body Mass Index (BMI). Body Mass Index (BMI) is a numerical value derived from the weight and height of an individual. It is widely used as a screening tool to categorize whether a person is at a healthy weight for their height.
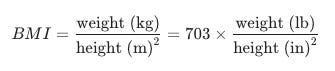
In the early 20th century, life insurance companies faced a problem: they needed a way to predict when people would die to set profitable premiums. They looked back at Adolphe Quetelet’s work and realized his Quetelet Index (the term “BMI” hadn’t yet been coined) was the most efficient way to categorize large groups of people. In 1942, Louis Dublin, a statistician for the life insurance company Metropolitan Life, used Quetelet’s concepts to create the first “Ideal Weight” tables. These tables weren’t based on how people looked, but on mortality data. In this care, “ideal” simply meant the weights associated with the lowest mortality rates among policyholders. To account for the variability that Quetelet’s math ignored, the industry invented the categories of “Small,” “Medium,” and “Large” frames, though they never actually provided a scientific way to measure someone’s frame.
By the 1950s, the insurance industry had data tables, but they lacked a clinical “seal of approval.” This is where Ancel Keys entered the picture. Keys was a physiologist famous for the Seven Countries Study, the first major study to examine how dietary patterns and lifestyle affect cardiovascular disease across different nations and cultures. He wanted to study heart disease but needed a simple way to measure largeness in thousands of participants without using expensive lab equipment. Keys tested various formulas against actual body fat measurements (using skinfold calipers and underwater weighing). He found that Quetelet’s 140-year-old index was the most accurate proxy for body fat percentage. In a landmark paper published in 1972, Keys officially dubbed the formula the Body Mass Index (BMI). He explicitly stated it was better than the insurance tables because it was purely mathematical and didn’t rely on the vague “frame sizes” the insurance companies used.
Nevertheless, most medical institutions in the first half of the 20th century viewed weight as a statistical risk factor, not a disease in itself. Medical consensus often mirrored societal bias, treating excess weight as a symptom of poor discipline or a deficit in character. Since the data came from insurance companies rather than biological labs, doctors often focused on the consequences (diabetes, heart disease) rather than the cause. If a patient didn’t have a secondary condition, they weren’t considered sick.
The shift toward viewing obesity as a disease was driven by a move from epidemiological observation (seeing that heavy people died sooner) to biological explanation (understanding why the body resists weight loss and how excess weight increases the risk of mortality). Large-scale studies provided the first undeniable link between excess weight and mortality. The Framingham Heart Study (FHS) was one of the most significant and longest-running epidemiological studies in medical history. The study set out to identify the common factors or characteristics that contribute to cardiovascular disease by following 5,209 adult residents of Framingham, Massachusetts who had not yet developed overt symptoms of heart disease or suffered a heart attack or stroke. By examining their bloodwork for over nearly 80 years and correlating it with clinical outcomes, researchers could pinpoint the risk factors that were associated with higher incidence of heart attack or stroke over the average person’s lifetime. The first official publication describing the Framingham Heart Study was published in March 1951 in the American Journal of Public Health. It was one of the first studies to prove that obesity was a direct, independent risk factor for cardiovascular disease. It showed that even if a person didn’t smoke or have high cholesterol, excess weight alone significantly shortened lifespan.
Nevertheless, there was a 34-year chasm between Framingham Heart Study’s first publication and recognition of obesity as a disease by major medical institutions. This occurred for a few reasons:
The belief that obesity would “fix itself”: In the 1960s and 70s, the medical focus was almost entirely on smoking cessation and managing blood pressure. Obesity was seen as a “secondary” concern that would naturally resolve if patients just followed the emerging (and now heavily scrutinized) “Food Pyramid” guidelines. There was no recognized medical specialty for obesity and most doctors felt unequipped to treat weight. They often viewed dieting as something that happened in the home or at commercial clubs like Weight Watchers (founded in 1963), not in a clinical setting.
The industrialization of food: The 1950s through the 1970s saw a massive shift in the American food landscape that outpaced medical policy. High fructose corn syrup (HFCS) began first entered the American food supply in 1967 by the Clinton Corn Processing Company. When the price of cane sugar skyrocketed in 1974 due to a global double-crop failure (European Beet Failure, Cuban Drought) and the sunsetting of the Sugar Act of 1937, manufacturers increasingly turned to HFCS. By the 1990s, HFCS was in almost everything, from bread and yogurt to salad dressings and infant formula. By 1999, the average American consumed roughly 63 pounds per year, up seven-fold from 9 pounds per year in 1977. As the food supply became increasingly industrialized, the average weight began to climb. Medical institutions initially responded with a “blame the patient” approach.
The absence of a biological mechanism: Until the mid-1980s, the prevailing scientific belief was that the human body was a simple “Calorie In, Calorie Out” machine. Without a clear biological mechanism to explain why some people struggled more with weight than others, there was no medical way to intervene.
By the mid-1980s, the pressure to recognize obesity became undeniable. The skyrocketing costs of treating Type 2 diabetes and heart disease (risk factors associated with obesity) began to threaten the stability of the healthcare system. Furthermore, thirty years of Framingham data and subsequent studies like the National Health and Nutrition Examination Survey (NHANES) suggested that the obesity epidemic was not an individual failure but a population-wide trend. NHANES II (1976–1980) showed a sudden and dramatic spike in the average American’s weight compared to NHANES I (1971–1974). This proved that the issue was not just a few “unmotivated” individuals, but a population-wide shift.
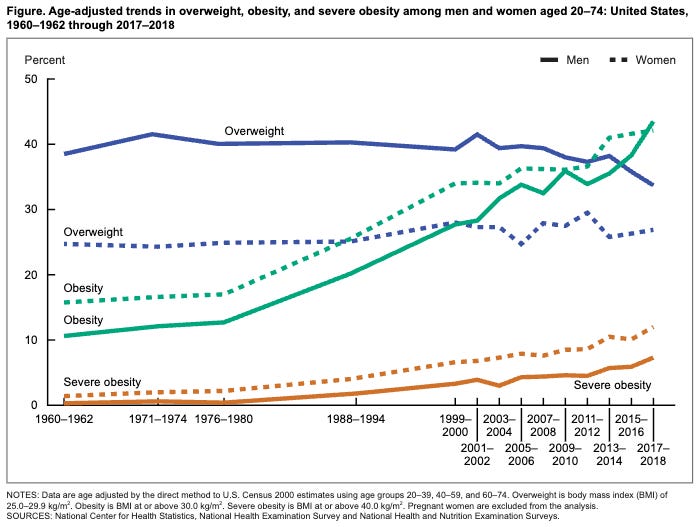
The National Institutes of Health (NIH) was one of the first major federal bodies to sound the alarm, though their initial stance was more about “risk” than “disease.” At the 1985 Consensus Conference, the NIH convened a panel that concluded obesity is a “disease” in the sense that it is a condition that impairs health. However, the report was cautious, focusing heavily on the association with other diseases like diabetes and hypertension.
Thirteen years later in 1998, the NIH released the first federal clinical guidelines based on BMI. By standardizing the BMI categories (25.0-29.9 for overweight; ≥30.0 for obesity), they effectively medicalized the condition for millions of Americans overnight. The NIH’s shift was driven by the desire to move clinical practice toward evidence-based weight management.
Despite the NIH’s shifting stance, the Medicare Coverage Issues Manual explicitly stated that “obesity is not considered a disease” until 2004, which acted as a total barrier to any form of treatment coverage. The change occurred, when the Centers for Medicare & Medicaid Services (CMS) officially removed that sentence from its manual. Following this, Medicare began covering certain intensive behavioral therapy (IBT) sessions and FDA-approved bariatric surgical procedures for patients meeting specific BMI and comorbidity thresholds. As of today (April 2026), Medicare still does not cover medications specifically for chronic weight management. This is not due to a clinical decision by CMS, but rather a statutory restriction. When the Medicare Part D prescription drug benefit was created in 2003, the law included a list of excluded drugs. This list specifically prohibited coverage for agents used for weight loss, anorexia, or weight gain. At the time, weight-loss drugs were often viewed more as “lifestyle” or “cosmetic” interventions rather than essential medicine. Medicare can cover these same medications if they are FDA-approved to treat a different condition that is not weight loss. For example, if a GLP-1 (Ozempic, Mounjaro) is prescribed to treat Type 2 diabetes or to reduce the risk of major adverse cardiovascular events (MACE) in adults with established heart disease, it may be covered.
The most significant turning point came in June 2013, when the American Medical Association (AMA) House of Delegates voted to recognize obesity as a disease. This was a rebellion against the AMA’s own Council on Science and Public Health. The Council actually recommended against the move, arguing that BMI was a flawed metric and that “disease” implies a specific pathology that wasn’t consistently present in everyone with a high BMI. Proponents argued that the willpower model had failed for decades. They insisted that by labeling it a disease, they would reduce social stigma, increase funding for research, and force insurance companies to cover treatments. The delegates overrode the council, officially declaring that obesity involves “multiple pathophysiological aspects” including genetic, environmental, and endocrine factors.
Once the AMA moved, other major global institutions followed, though with varying degrees of nuance. In 2017, the World Obesity Federation pushed the definition further, arguing that obesity fits all the criteria of a disease: it has a known cause (energy imbalance), characteristic signs/symptoms, and leads to harm. In 2021, the European Commission finally recognized obesity as a “chronic relapsing disease,” which paved the way for better access to care across the EU.
Even with these recognitions, the debate remains alive in the medical community today around two main points:
The Flaw of BMI: Many doctors argue that using BMI as the sole diagnostic tool for a “disease” is scientifically unsound, as it ignores muscle mass and metabolic health.
The Stigma Paradox: Some advocates worry that the “disease” label takes away a person’s sense of agency, while others argue it is the only way to get the medical system to take the condition, and the patient, seriously.
Hormones In, Hormones Out
The transformation of obesity from a lifestyle issue to a biological disease was driven by the discovery of an invisible signaling system, a hormonal conversation between the stomach, fat tissue, and the brain. For decades, we believed the body was like a bucket; if you poured in more calories than you leaked out, you got heavier (“calories in, calories out”). These discoveries proved the body actually has a biological thermostat that fights to maintain a body weight set point.
It all started in 1949 at the Jackson Laboratory in Bar Harbor, Maine, the world’s premier center for mouse genetics. A research technician named Margaret Dickie was working in a colony of mice when she noticed something bizarre. Two siblings in a litter were rapidly becoming much larger than their cage mates. By the time these mice were a few weeks old, they were roughly three times the weight of their normal siblings. They were round, lethargic, and possessed an insatiable appetite. Researchers realized this was a spontaneous genetic mutation because the trait only appeared if both parents carried the hidden gene. The scientists used cross-breeding experiments to determine that it was a recessive mutation (mice needed both copies of the gene to have the obese phenotype) and they labeled the hypothetical gene as ob (for obese).
Before this accident, scientists had no reliable way to study the biology of weight. Humans were too genetically diverse and their environments were too varied to control for. The ob/ob mouse provided two valuable insights:
Genetic Uniformity: Every ob/ob mouse was genetically identical, allowing researchers to isolate weight as the only variable.
A Biological Driver: It proved that an error in the genetic code could override all other behaviors. These mice weren’t choosing to overeat, so much as their biology was forcing them to.
While the mice were discovered in 1949, the mechanism by which mutations in the ob gene caused obesity remained a mystery for nearly half a century. Scientists spent decades trying to figure out what that single ob gene was supposed to do. Some thought the mice had a slow metabolism; others thought their stomachs were simply larger.
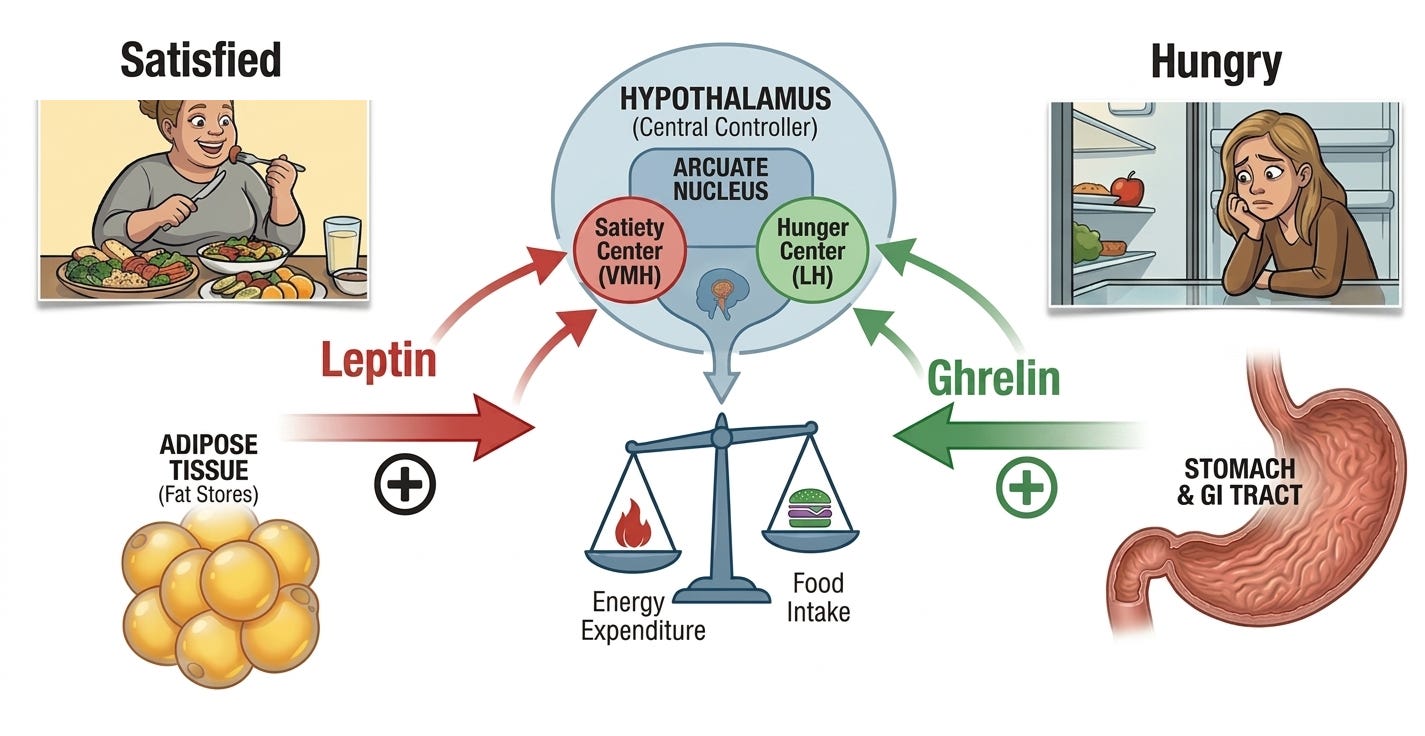
It wasn’t until Douglas Coleman’s parabiosis experiments in the 1960s that researchers realized the ob/ob mice were missing a signal in their blood. In these parabiosis experiments, Coleman physically joined the circulatory systems of the ob/ob mice to a healthy mouse. He noticed that the ob/ob mouse stopped overeating and lost weight. This proved that the healthy mouse was sending a satiety factor through the blood that the ob/ob mouse was missing.
In 1986, Dr. Jeffrey Friedman at Rockefeller University set out to find the specific gene responsible for the ob/ob mutation. He used a technique called positional cloning, which is essentially looking for a needle in a haystack by slowly narrowing down the location on the chromosome. Dr. Friedman’s team spent eight years mapping the mouse genome. They slowly narrowed the search from an entire chromosome down to a specific region containing several hundred genes. In 1994, they identified a gene that was expressed only in adipose tissue (fat cells). In the ob/ob mice, this gene was mutated and broken, meaning they couldn’t produce the protein it coded for. Once the gene was isolated, Dr. Friedman’s team used it to produce the protein in a lab. They named it leptin, from the Greek word leptos, meaning “thin.” When they injected the synthetic leptin protein back into the ob/ob mice, the effect was staggering. The mice, which had previously been obsessed with food, suddenly lost their appetite. Their metabolic rates increased, and they shed the excess fat rapidly. Dr. Friedman then showed that humans also have the ob gene and produce leptin (LEP). This proved that the weight-regulation system seen in mice was a fundamental part of human biology.
Dr. Friedman’s discovery shattered the thermodynamic view of obesity that more “calories in” than “calories out” was an easy goal to accomplish through sheer force of will. Instead, obesity was in part a neuroendocrine disease that results from broken hormone signaling. His findings established three scientific pillars:
Adipose is an Endocrine Organ: It proved fat isn’t just storage; it is a vital organ that talks to the brain.
The Biological Set Point: It explained why dieting is so hard. When you lose fat, your leptin levels drop, and your brain thinks you are starving, triggering intense hunger to return you to your body weight set point.
Leptin Resistance: While Friedman initially hoped leptin would be a treatment, he discovered that most people with obesity have high leptin levels. Their brains have become resistant to the signal, much like a type 2 diabetic is resistant to insulin.
Dr. Friedman’s cloning of the gene encoding leptin (LEP) in 1994 triggered a flurry of activity in obesity genetics, where researchers used positional cloning, a tedious process of mapping where a a gene’s location on a chromosome without knowing what the gene actually does. In 1995, researchers cloned the leptin receptor gene (LEPR) responsible for the db/db mouse, proving it was the receiver for the leptin signal. In 1997, using candidate gene approaches (looking at genes already known to be involved in brain signaling), researchers identified mutations in children with severe obesity, red hair, and adrenal insufficiency (POMC) or severe digestive issues (PCSK1). The following year in 1998, scientist identified the gene encoding the Melanocortin-4 receptor (MC4R) as the end-point for satiety signals in the brain. When they screened patients with extreme early-onset obesity, they found the mutations.
While Dr. Jeffrey Friedman’s 1994 discovery of leptin identified the “brakes” of the energy system, the scientific community was still hunting for the “gas pedal.” In 1999, Dr. Masayasu Kojima and his team at the Kurume University School of Medicine in Japan finally identified ghrelin, the first (and only) known hormone that signals the brain to start eating. Kojima’s discovery was a masterclass in reverse pharmacology, relying heavily on synthetic molecules created by pharmaceutical companies years earlier.
The story starts with scientists trying to create a better medicine, not trying to find a new hormone. In the late 1970s, researchers at the pharmaceutical company Merck were experimenting with small, synthetic molecules to see if they could trigger the release of Growth Hormone (GH). They eventually created a class of drugs called Growth Hormone Secretagogues (GHS). These worked incredibly well in the lab, but they were entirely man-made; they didn’t exist in nature. By the mid-90s, gene-mapping technology had advanced significantly. Scientists at Merck, led by Andrew Howard, decided to work backward. They took these synthetic GHS drugs and used them as bait to see what protein they were sticking to in the brain. They successfully isolated a specific protein on the surface of cells in the pituitary gland and hypothalamus, two regions in the brain. When the synthetic drug contacted this protein, the cell reacted. They found the gene that coded for this protein and named it the GHS Receptor (GHS-R).
At this point, they had a receptor that responded perfectly to a synthetic hormone (the Merck drugs). However, evolution doesn’t build receptors for synthetic hormones. This meant there had to be a natural, biological hormone already inside the human body that usually turned that lock. In biology, a receptor with no known natural partner is called an orphan receptor. From 1996 to 1999, the GHS-R was one of the most famous orphan receptors in science. Everyone knew it was there, and they knew it probably controlled hunger and growth, but they couldn’t find the source of the hunger signal.
This is where Dr. Masayasu Kojima stepped in. Instead of looking for a hormone and then finding what it did, he used the receptor as a hook and went fishing for the hormone. He created a tester cell line that had the GHS-R receptor on its surface and a calcium-sensitive fluorescent dye. If any substance touched the receptor and activated it, the cell would immediately release calcium and glow. He then took crude extracts from various rat tissues (stomach, brain, heart, etc.) and poured them over these tester cells. When he poured the extract from the stomach, the cells lit up. To isolate the specific hormone, Dr. Kojima used High-Performance Liquid Chromatography (HPLC) to filter the stomach extract into so-called fractions, different mixtures containing molecules with different sizes, charges, and hydrophobicites (how much they repel water). He repeated this process over and over, changing the chemical conditions each time, until he had a single, pure peak on his mass spectrometry graph, a single protein substance that consistently activated the GHS-R receptor. That molecule turned out to be a 28-amino acid peptide that he named ghrelin. Dr. Kojima even identified a unique structural characteristic of the stomach-derived hormone. His mass spectrometry graph showed a mystery mass attached to the third amino acid in the chain. He realized a fatty acid called n-octanoic acid was hanging off the side of the hormone. This fatty acid tail is what allows ghrelin to cross the blood-brain barrier and hit the receptor. Without that specific oily tail, the hormone would be locked out of the brain and never reach its receptor.
Dr. Masayasu Kojima and his colleagues published their groundbreaking discovery of ghrelin in the prestigious scientific journal Nature on December 9, 1999. The paper was revolutionary because it fundamentally changed how we understand the gut-brain axis. The key takeaways included:
The First Hunger Hormone: It identified the first (and still only) known hormone produced in the periphery (the stomach) that travels to the brain to directly stimulate hunger.
The Orphan Solution: It solved the 3-year mystery of the GHS Receptor (the “lock”) by providing its natural “key.”
Chemical Uniqueness: It revealed that ghrelin requires a specific fatty acid (n-octanoic acid) to function, a chemical modification that was previously unheard of in mammalian hormones.
Revealed the Enteroendocrine System: It proved that the stomach, an organ previously thought of only for digestion, was actually a critical part of the endocrine system, regulating both growth hormone levels and energy balance.
The publication of this paper is often cited as the definitive end of the willpower-only era of weight management, providing the final biological proof needed for medical institutions to begin categorizing obesity as a complex metabolic disease.
The next quantum leap forward in obesity genetics was ushered in by the Human Genome Project, an international scientific research collaboration aimed at mapping the DNA sequence of the average human. The identification of the FTO gene was the first major success of the Genome-Wide Association Study (GWAS), a research approach that correlates genetic variations (genotype) with a particular disease or trait (phenotype). Researchers scanned the DNA of 38,000 people. They didn’t have a guess or a candidate, the computer simply flagged a specific region on Chromosome 16 (FTO) that was consistently different in people with higher BMIs. As computers got faster and biobanks (like the UK Biobank) grew to 500,000+ people, researchers discovered TMEM18, NEGR1, and over 1,000 other loci. These were found by comparing the full genomes of hundreds of thousands of people to find the signal in the noise.
Today, genetic obesity is typically categorized by how hard-coded the weight gain is. As of 2026, we categorize these genes into three distinct buckets:
Monogenic (single-gene)
Syndromic (part of a larger syndrome)
Polygenic (thousands of small variants)
Monogenic obesity is caused by mutations in one gene in the Leptin-Melanocortin Pathway. These are rare, highly penetrant mutations. If a person has a defect in these genes, severe, early-onset obesity is almost guaranteed, regardless of diet or exercise.
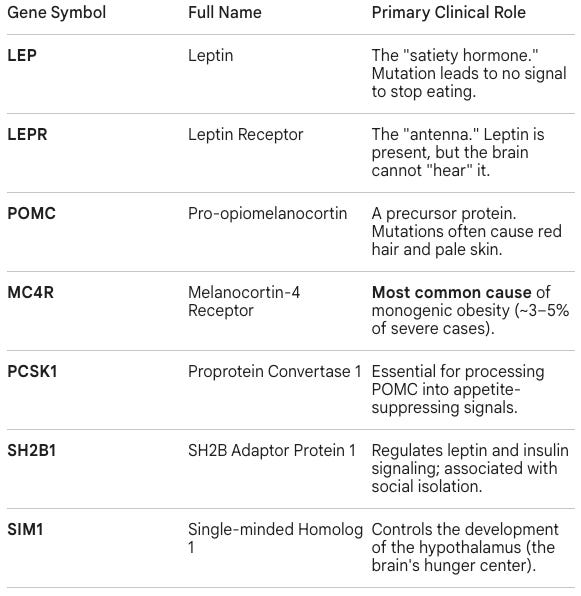
Syndromic obesity is caused by mutations that lead to multi-system disorders. In these cases, obesity is a hallmark feature, but it is accompanied by other symptoms like intellectual disability, vision loss, or organ malformation. They include:
PWS (Prader-Willi Syndrome): Caused by a loss of expression in the 15q11.2–q13 region on the paternal chromosome. The loss of these paternal genes causes structural and functional defects in the hypothalamus. In a healthy brain, the hypothalamus balances “hunger” signals (AgRP/NPY neurons) and “satiety” signals (POMC neurons). The loss of key genes in PWS patients cause the brain to behave as if the body is in a state of constant, severe starvation. The condition is famously associated with hyperphagia, an insatiable and life-threatening hunger.
BBS (Bardet-Biedl Syndrome): Linked to mutations in at least 21 genes (e.g., BBS1, BBS10). It is a ciliopathy, where the tiny hairs on cells (cilia) in the hypothalamus (the brain’s appetite control center) can’t sense hormones properly.
Albright’s Hereditary Osteodystrophy: Mutations in the GNAS gene leads to obesity, short stature, and bone formation in unusual places.
Polygenic obesity accounts for relatively less severe phenotypes that don’t cause obesity in most cases, but nevertheless increase the susceptibility to developing obesity. These were largely discovered through the GWAS studies and include:
FTO (The “Fat Mass” Gene): The most famous GWAS hit. It influences whether your body stores energy as white fat or burns it as beige/brown fat.
MC4R (Common Variants): While rare mutations cause monogenic obesity, common small variations in MC4R contribute to the weight of millions of people.
TMEM18 & NEGR1: Highly expressed in the brain; they influence synaptic plasticity and how your brain “wires” its hunger cues.
KCNMA1: A more recent addition to the GWAS catalog (highlighted in 2026 data) that influences neuronal excitability and energy intake.
While these genetic factors teach us how body weight set points work, <5% of cases of obesity have monogenic & syndromic roots. The vast majority of obesity cases, the remaining 95%, are polygenic. This means there isn’t one broken gene, but rather hundreds or thousands of tiny variations (SNPs) that cumulatively increase a person’s risk of obesity. While genetics accounts for one contributing factor for how our bodies manage energy, the disease is typically a mixture of factors, including biological predispositions, environmental influences (such as the availability of ultra-processed foods), socioeconomic conditions, and lifestyle habits.
Conclusion
The history of obesity is a narrative of shifting mirrors. For centuries, we looked at the scale and saw a reflection of our character, our status, or our self-discipline. But as we have moved from the artistic curves of the Renaissance to the precise mapping of hormones the regulate hunger and obesity-related genes, that mirror has been replaced by a microscope. The discovery of leptin and ghrelin didn’t just provide us with new biological vocabulary; it fundamentally reclassified obesity as a neuroendocrine struggle, a war between a modern environment and an ancient, survival-focused thermostat. We now know that for many, the body weight set point is less of a choice, and more of a genetic and hormonal blueprint.
However, understanding the biology was only half the battle. While science spent the late 20th century uncovering the ‘why’ of weight, the medical world was simultaneously struggling with the ‘how’ of treatment, navigating a dark age of dangerous pills and failed interventions.
Stay tuned for Part 2, where we explore the evolution of obesity treatments and how the field climbed out of a century-long dark age and emerged into the current golden age of GLP-1s.
To contact us, please send us an email at biotechreadout@gmail.com
Disclaimers
Investigational Status Disclaimer
The therapeutic candidates discussed in this newsletter are currently in clinical development and have not been approved for commercial sale by the U.S. Food and Drug Administration (FDA), the European Medicines Agency (EMA), or other global regulatory authorities. Their safety and efficacy have not been established. References to pipeline products and ongoing clinical trials involve significant risks and uncertainties. Statements regarding the potential safety, potency, or efficacy of investigational drugs reflect current hypotheses and are not a guarantee of future performance or regulatory clearance. The outcome of clinical trials is inherently unpredictable, and clinical results from earlier stages may not be predictive of results in later, larger-scale trials.
No Medical Advice Disclaimer
This newsletter is for informational and educational purposes only. The content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or another qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read in this publication.
No Patient-Provider Relationship Disclaimer
The information provided in this newsletter is for educational and analytical purposes only. Receipt of this information, or any interaction with this content, does not create a physician-patient, pharmacist-patient, or any other professional-provider relationship between you and the authors or publishers. This newsletter should not be used as a substitute for a personal consultation with a qualified healthcare professional.
Forward-Looking Statements Disclaimer
This newsletter contains “forward-looking statements” regarding future events, including clinical trial timing, regulatory milestones, and projected market performance. These statements are based on current expectations and assumptions that are subject to significant risks and uncertainties. Actual results may differ materially from those expressed or implied. We undertake no obligation to update these statements as a result of new information or future developments.
Third-Party Links & Content Disclaimer
This newsletter contains links to third-party websites, including clinical trial registries and corporate presentations. Biotech Readout does not endorse, guarantee, or assume responsibility for the accuracy or reliability of any information offered by third-party providers.
Errors and Omissions Disclaimer
While we strive for technical accuracy, the information in this newsletter is provided on an “as is” basis with no guarantees of completeness, accuracy, or timeliness. Biotech Readout assumes no liability for any errors or omissions in the content of this publication.
Non-Endorsement Disclaimer
Any reference to specific commercial products, processes, or services by trade name, trademark, or manufacturer does not constitute or imply an endorsement or recommendation by the author. All trademarks are the property of their respective owners.
No Investment Advice Disclaimer
This newsletter is for informational purposes only and does not constitute financial, investment, or legal advice. The author is not a registered investment advisor. You should consult with a professional financial advisor before making any investment decisions. The biotechnology sector is highly volatile; past performance is not indicative of future results.
Conflict of Interest Disclaimer
The author of this newsletter maintains a position of independence. At the time of publication, the author holds no direct financial interest, equity, or options in any of the companies mentioned in this report. No compensation has been received from any third party to feature or analyze specific therapeutic candidates or corporate entities.