
Hepatitis C, Part 1
How Schering-Plough, Roche, Merck, Vertex, and Janssen turned the tide from a public health epidemic to an 80% cure rate

Disclaimer: This newsletter is for educational and informational purposes only and does not constitute medical, investment, or financial advice, nor does it establish a provider-patient relationship. Content may include forward-looking statements and discussions of investigational therapeutic candidates that are not FDA/EMA approved; their safety and efficacy remain unestablished and clinical outcomes are unpredictable. While we strive for accuracy, all information is provided as is without guarantees. As of the date of publication, the author holds no direct equity positions in the specific companies mentioned in this issue nor receives third-party compensation for this coverage. Please find a complete version of our disclaimers at the bottom of this article and on our About page.

Prologue
In Part 1 of this two-part series on Hepatitis C, we dive into the Nobel-prize winning discovery of Hepatitis C and explore how Schering-Plough, Roche, Merck, Vertex, and Janssen turned the tide from a public health epidemic to an 80% cure rate.
Subscribe so you don’t miss Part 2, where we explore how Gilead bested big pharma in the race for a cure.
Not A, Not B
The story of hepatitis starts in the theater of war. For centuries, an infectious form of jaundice was observed in military campaigns and crowded, unsanitary living conditions. Those who contracted it exhibited characteristic yellowing of their skin and the whites of their eyes. At the time, catarrhal jaundice, as it was then called, was believed by many to be a mechanical issue, specifically a blockage of the bile duct by mucus. However, the sheer scale of outbreaks in military camps and the clear patterns of spread suggested an infectious pathogen was responsible. It was only in the 1940s when the desperate circumstances of World War II spurred scientists into action. Dr. W. Paul Havens and his team at Yale University were tasked with proving that catarrhal jaundice was a transmissible, infectious disease, even without the ability to see the virus under the microscopes of the era. To prove the mode of transmission, Havens and his team turned to controlled human experiments. In these studies, they worked with volunteer subjects, a common, though now deeply controversial, practice in the mid-20th century.
The researchers collected samples of blood serum and stool from patients who were suffering from acute cases of infectious hepatitis. These samples were processed to create filtrates, liquids that had been passed through fine filters to remove bacteria, leaving only the smaller, unidentified viral agents. In a series of trials, these filtrates were administered to volunteers via feeding or injection. By observing the volunteers over several weeks, the researchers were able to track who developed the classic symptoms of hepatitis, such as jaundice, fever, and liver enzyme elevation. Through these experiments, Havens successfully demonstrated that what we now know as Hepatitis A was primarily transmitted via the fecal-oral route. He showed that the agent was present in the stool of infected individuals, proving that contaminated food & water or poor sanitation were the primary engines of the disease’s spread. One of Havens’ most crucial contributions was his ability to clearly differentiate “infectious hepatitis” (Hepatitis A) from “serum hepatitis” (Hepatitis B) by comparing the incubation periods and transmission routes. his team observed two distinct patterns:
Hepatitis A (Infectious): Had a relatively short incubation period and was easily transmissible through the ingestion of contaminated stool or serum.
Hepatitis B (Serum): Had a much longer, more insidious incubation period and was primarily linked to the injection of blood or plasma products, such as the contaminated yellow fever vaccines that had caused massive outbreaks in the military.
These findings were instrumental in the wartime effort. By identifying the fecal-oral route of Hepatitis A, military leaders could finally implement effective sanitation protocols and food-handling safeguards to curb outbreaks. Beyond the immediate tactical gains, Havens’ research laid the essential groundwork for the future of virology. In 1973, a team led by Dr. Stephen Feinstone at the National Institutes of Health (NIH) used a technique called immune electron microscopy. They took samples from volunteers who had been infected with the virus and mixed them with serum from patients who had recovered from the illness. Under the powerful magnification of an electron microscope, they observed the antibodies from the recovered patients clumping together around tiny, circular particles. These particles were the Hepatitis A virus (HAV). By finally visualizing the virus, researchers were able to confirm its structure, paving the way for the development of a highly effective vaccine that would eventually make the disease largely preventable.
But, how could physicians diagnose a patient with Hepatitis A or B, aside from crude observations of symptoms? Dr. Baruch Blumberg, a researcher at the NIH, would stumble into that question and subsequently provide the answer. He was not initially looking for a hepatitis virus. Instead, he was studying human genetic diversity, specifically looking at how different populations responded to blood transfusions. Dr. Blumberg was analyzing the blood of diverse groups, including Indigenous Australians and hemophiliacs, to see how their antibodies reacted to foreign proteins. In 1963, Blumberg discovered an unusual reaction: the blood of a hemophiliac patient reacted with an antibody found in the blood of an Australian Aboriginal person. He dubbed this mysterious protein the Australia Antigen.
For several years, Blumberg worked to identify what this antigen actually was. In a profound realization in 1967, he linked the presence of this antigen directly to patients suffering from serum hepatitis (Hepatitis B). He had accidentally stumbled upon a surface protein of the virus, and the name of the Australia Antigen was changed to the more appropriate Hepatitis B surface antigen (HbSAg). This discovery was revolutionary. It meant that for the first time, scientists could test blood for the presence of this antigen, effectively creating the first screening test to keep the blood supply safe from Hepatitis B. For his persistent detective work in identifying this new virus, Baruch Blumberg was awarded the Nobel Prize in Physiology or Medicine in 1976.
By the mid-20th century, Hepatitis A and Hepatitis B were well understood by the medical community. Researchers had developed vaccines and screening tests for these viruses, which significantly improved the safety of the blood supply. However, by the 1970s, a frustrating trend emerged: a significant number of patients who received blood transfusions continued to develop hepatitis, even though their blood had been meticulously screened for both Hepatitis A and B. Clinicians were baffled. If the two known culprits were ruled out, what was causing this transfusion-associated hepatitis? For nearly 15 years, the medical community was forced to label this mysterious, stealthy pathogen as Non-A, Non-B hepatitis. It was deadly and chronic, leading to liver cirrhosis and cancer, but invisible to the tests of the time.
Dr. Harvey J. Alter, working at the National Institutes of Health (NIH), took on the role of investigating this illusive disease. He started by observing the patterns of Non-A, Non-B hepatitis in patients. Similar to the discovery of Hepatitis A & B, Alter demonstrated that blood from patients with this mysterious hepatitis could transmit the disease to chimpanzees, the only animal model susceptible to it at the time. Crucially, Alter’s experiments showed that the agent was a virus. He and his team discovered that the infectious material could be filtered in a way that captured viral particles but excluded bacteria. It was a breakthrough, but the virus was present in such incredibly low concentrations that it remained impossible to capture or see. It also refused to grow in any laboratory cell culture. For over a decade, Alter’s work hit a wall.
In 1989, Dr. Michael Houghton and his colleagues Qui-Lim Choo and George Kuo at the Chiron Corporation picked up where Alter left off, but they took a radically different approach. Instead of trying to grow the virus on petri dishes, they searched for its genetic footprint using shotgun sequencing. Shotgun sequencing is a laboratory method used to determine the entire DNA sequence of an organism’s genome. Instead of reading a long DNA strand from start to finish, the process shatters the genome into millions of smaller fragments (“shotgun“). Each of these small fragments is sequenced independently using automated technology to determine the order of its chemical bases (A, T, C, and G). Powerful computer algorithms close the loop by looking for overlapping sections to figure out how they fit back together. Like assembling a giant jigsaw puzzle without the original picture, the computer matches the overlapping edges to reconstruct the original, long, continuous sequence of the entire genome.
The Chiron team started with a massive amount of serum taken from a chimpanzee that had been infected with Non-A, Non-B hepatitis. They extracted all the genetic material (RNA) from the serum and converted it into DNA. They then inserted these millions of DNA fragments into bacteria, creating a library of clones. This library contained genetic material from the chimpanzee, the bacteria, and, hopefully, the hidden virus. The team had to figure out which clone belonged to the virus. The team created an innovative probe. They took serum from a patient who was known to have Non-A, Non-B hepatitis, reasoning that this patient must have developed some antibodies against the virus, even if they were too weak to detect by standard means. They used this serum to scan the millions of clones in their library, looking for any interaction between the patient’s antibodies and the bacterial clones. For years, the team screened millions of clones with no luck. Finally, in 1989, they hit upon a clone, which they labeled “5-1-1.” It reacted with the patient’s antibodies. When they sequenced the DNA of this clone, they confirmed it was not from the chimpanzee or the bacteria. It was a piece of the viral genome.
Once they had the virus’ genetic sequence, the impact was immediate and profound. Within months, Chiron developed a blood test that allowed screening centers to identify and remove virus-contaminated blood from the supply, virtually eliminating transfusion-transmitted infections. They officially named the virus Hepatitis C. For his decade of tireless, innovative work Michael Houghton was jointly awarded the 2020 Nobel Prize in Physiology or Medicine, alongside Harvey J. Alter and Charles M. Rice.
Panning for Medicines
For those who aren’t familiar with the California Gold Rush, it was a transformative event in American history that turned a quiet, remote Mexican territory into a bustling state almost overnight. It began on January 24, 1848, when James Marshall, working for Swiss immigrant John Sutter, discovered gold flecks in the tailrace of a sawmill being built in Coloma. While the news was initially met with skepticism, it quickly ignited a global migration. By 1849, Forty-Niners were flooding into California from across the U.S., Europe, China, and South America. The sudden influx of hundreds of thousands of people resulted in the rapid development of cities like San Francisco and Sacramento, as prospectors harvested gold nuggets worth tens of billions of dollars.

Much like the California Gold Rush, the discovery of a diagnostic for Hepatitis C in the early 1990s lit a fuse that ended in the explosive realization of how large the unmet need truly was. It is estimated that in the 1970s and 1980s, about 4% of patients who received a blood transfusion contracted Hepatitis C. Some reports say that the risk was as high as 10% for recipients of blood transfusions in some settings. Since the virus had been invisible for so long, millions of people were unknowingly infected through medical procedures, blood products, or injection drug use years or decades prior. Epidemiologists at the time began to realize that they were standing at the base of a coming tsunami of potentially fatal end-stage liver disease. Since the virus can remain asymptomatic for 20 to 30 years, the surge in liver cancer and cirrhosis cases seen in the 1990s was actually the result of infections contracted in the 1960s and 1970s. This meant that even if new infections were stopped instantly, the public health burden of the disease would continue to climb for decades. It was a chance for biotech and pharma to make a substantial impact on health outcomes by bringing safe and effective medicines to market, and it would soon become the talk of the town amongst drug hunters.
Of course, the Gold Rush also had its cast of characters. The most successful underdog was, arguably, Samuel Brannan. He was a Mormon businessman who, like many, moved to California. Unlike many, he did not dig for gold. Brannan famously ran through the streets of San Francisco waving a bottle of gold dust to announce the discovery, knowing his stores were perfectly positioned to supply the incoming tide of prospectors. By buying up all the mining supplies in the region and reselling them at massive markups, he became California’s first millionaire. Samuel Brannan reminds us that a small, but scrappy player can outmaneuver established giants that are initially thought to be in the best position to capitalize on a new trend. One such established giant was John Sutter, the owner of the land where gold was first discovered in California. Instead of being the ultimate winner, the Gold Rush destroyed his agricultural empire. Prospectors overran his land, squatters seized his property, and his workers abandoned him to search for gold. He died a man of modest means, having lost his “Nuevo Helvetia” empire to the very event he inadvertently started. James Marshall, the man who actually spotted the gold at the mill never realized a fortune from it either. Like his employer Sutter, his claim was overrun by others, and he spent the later years of his life in obscurity, unable to capitalize on his own discovery.
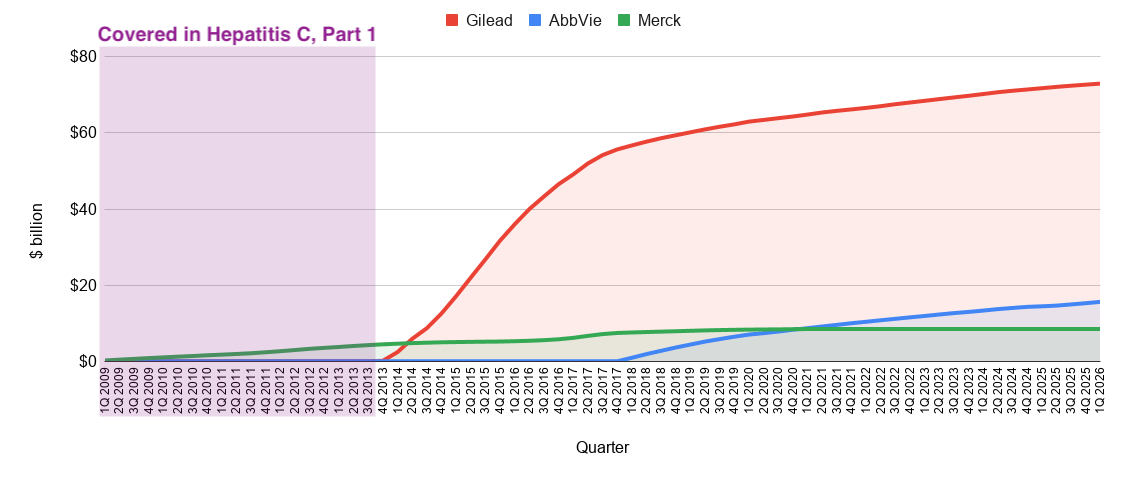
The race for the Hepatitis C cure has a similar cast of characters. Unlike, the folly of the established giants in the Gold Rush, the giants in the Hepatitis C race would still end up with blockbusters on their hands. So, no hard feelings. However, it was a hard fought battle against Gilead, a scrappy pioneer in the field of virology, but an underdog nonetheless. Gilead had earned its keep with its HIV/AIDS franchise. When the race truly kicked off in 2010, the company was generating annual revenues of around $8 billion. As a traditional big pharma powerhouse, Merck was significantly larger, with 2010 annual revenues exceeding $45 billion. It had a much broader portfolio spanning various therapeutic areas, including vaccines, oncology, and cardiovascular health. They also had a 16-year head start in Hepatitis C compared to Gilead. Abbott Laboratories was another a massive, diversified giant, with revenues in the range of $35 billion to $40 billion in 2010. At the time, it included a vast array of medical devices, diagnostics, and branded pharmaceutical products (the latter of which would be spun off into AbbVie in 2013). Most importantly, AbbVie was ramping sales of the soon-to-be best selling drug of all time, Humira, giving them a formidable war-chest for cracking whichever market they decided to compete in.

Of course, how could we forget about the prospectors? The prospectors of the California Gold Rush were a diverse group of people driven by the “Gold Fever”. However, their lived experience was far removed from the romanticized image of striking it rich overnight. Prospecting involved standing in icy mountain streams for 10 to 12 hours a day, manual labor that led to chronic rheumatism, pneumonia, and exhaustion. While a tiny minority returned home with generational wealth, the vast majority found only marginal earnings and headed back home. Much like the prospectors of the Gold Rush, the prospectors of the Hepatitis C race waded into the icy competition long enough to get an FDA-approved product across the line. Rebuffed by the breakneck speed of innovation, they ultimately bowed out of the race and returned to the comfort of their core franchises.

The Interferon Incumbent
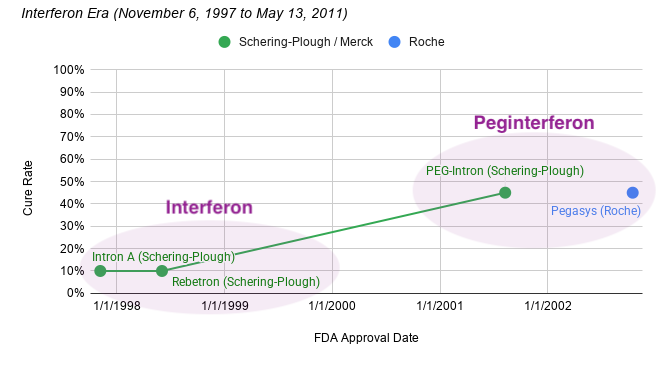
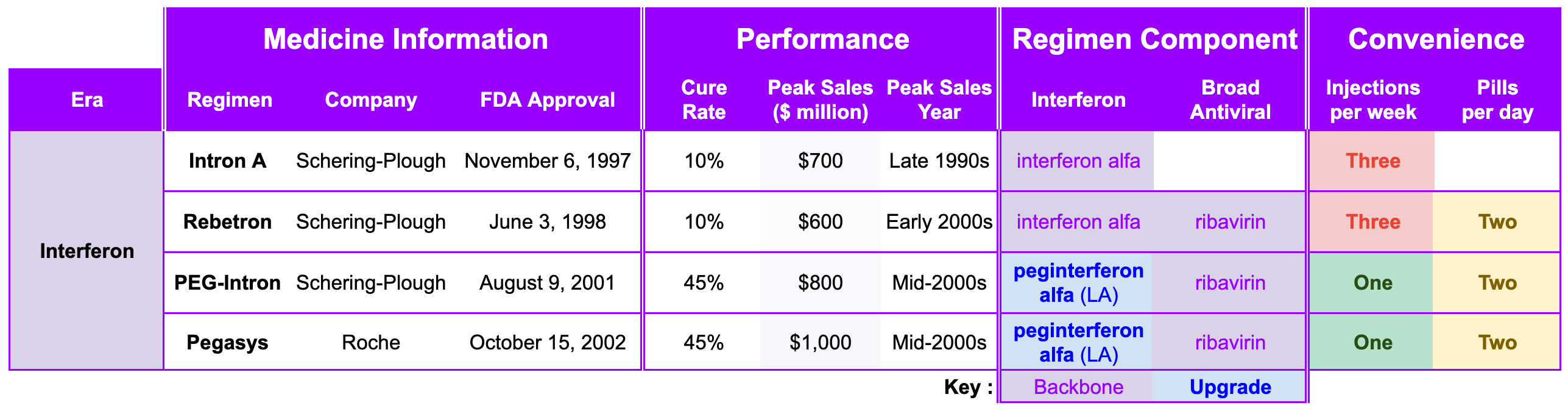
James Marshall discovered the first gold flecks in the California Gold Rush, but who was the “James Marshall of Hepatitis C”? The story begins in 1957 at the National Institute for Medical Research (NIMR) in Mill Hill, London. Two virologists, Alick Isaacs and Jean Lindenmann, were studying how cells fought off influenza viruses. Alick Isaacs was a Scottish physician and microbiologist and served as the head of the World Influenza Centre at NIMR. His counterpart, Jean Lindenmann, was a Swiss virologist and immunologist who had joined Isaacs at the NIMR in 1956. They discovered that when cells were exposed to an inactivated virus, they released a protein that seemed to “interfere” with the ability of other viruses to infect surrounding cells. They named this miraculous, protective protein interferon.
For years, interferon remained a laboratory curiosity because it was incredibly difficult and expensive to harvest from human cells in sufficient quantities. However, with the advent of recombinant DNA technology in the 1970s and 80s, the very same technology that would later allow for the mapping of the Hepatitis C genome, scientists finally learned how to manufacture human interferon on an industrial scale.
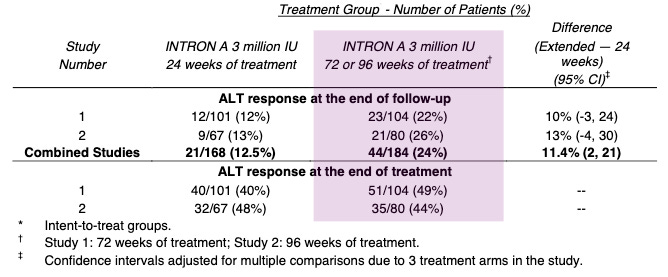
When Hepatitis C was finally unmasked in 1989, the medical community was desperate for a weapon. The virus was causing chronic liver disease in millions, and the options for treatment were nonexistent. Schering-Plough, a pharmaceutical company that had heavily invested in interferon research, recognized that this protein could be the key to attacking the newly identified virus. When Intron A (recombinant interferon alfa-2b) was under development and review for hepatitis C, the clinical trials focused on assessing its ability to normalize serum aminotransferases (ALT), which was used as a surrogate biomarker for reducing liver inflammation. Across the combined Phase 3 studies, patients receiving extended-term therapy (72 or 96 weeks) achieved a 24% sustained ALT response rate, compared to 12.5% for those treated for only 24 weeks. This represents a statistically significant improvement of 11.4% (95% CI: 2, 21) for the extended treatment group. At the end of the treatment period (when the drug is still active in the patient’s system), response rates were 40-48% for the 24-week group and 44-49% for the extended treatment group. On November 6, 1997, the FDA approved Intron A, the first therapy for Hepatitis C. It was a milestone moment: for the first time, there was a drug that could potentially slow the progression of the virus.
While interferon was a revolutionary discovery, its use as a Hepatitis C treatment was fraught with immense difficulty. Intron A required three injections per week, and in later years, as combinations with ribavirin were introduced (Rebetron approved on June 3, 1998), the physical toll became even more apparent. Patients often described the side effects as feeling like having a perpetual, debilitating flu. Symptoms included severe fatigue, depression, high fevers, and body aches. Since the treatment had to be taken for up to 48 weeks for optimal effect, many patients found it nearly impossible to complete. Cures could be achieved with interferon, but it was rare. In Hepatitis C, a cure rate refers to the percentage of patients who successfully clear the virus from their bodies completely after finishing treatment. A blood test must confirm the virus is undetectable 12 or 24 weeks after stopping all treatment (SVR12 or SVR24), in order to meet the medical criteria for a cure. A cured patient will still test positive for Hepatitis C antibodies, but the actual virus should be below the lower limit of detection. In the early years of interferon, the cure rate for interferon-based therapy was often less than 10%.
One of the primary issues with interferon was its short half-life in the body. Since the drug was cleared from the system quickly, patients had to endure the physical and emotional burden of three injections per week. Even with this frequent dosing, the drug remained in the bloodstream only for a short time, which limited its ability to maintain a consistent antiviral effect. Schering-Plough addressed this limitation through a process called pegylation. By chemically attaching a large, inert molecule called polyethylene glycol (PEG) to the interferon molecule, Roche created a shield that:
Extended Half-Life: Significantly slowed the absorption of the drug and its clearance from the body.
Enabled Once-Weekly Injections: Allowed the drug to remain at therapeutic levels in the bloodstream for an entire week, enabling a transition from three injections per week to just one.
Improved Efficacy: The more consistent drug levels achieved with once-weekly dosing led to higher sustained virologic response (SVR) rates compared to the older, non-pegylated interferons.
Schering-Plough refined the drug into pegylated interferon and PEG-Intron approved August 9, 2001). Two months later, Swiss pharmaceutical company Roche (a prospector) introduced their own pegylated interferon called Pegasys to the market on October 15, 2002. However, cure rates stubbornly remained at 40-50% for about a decade. Despite the severe side effects and limited efficacy, Schering-Plough’s pioneering work with interferon established a powerful precedent for Hepatitis C therapy, showing that the virus was susceptible to antiviral intervention. They also created the clinical infrastructure that would eventually bridge the gap to the modern era (diagnostic testing, patient monitoring, and specialized treatment centers).
The ensuing decade saw a meaningful consolidation of the pharmaceutical industry (and the Hepatitis C landscape), due to the loss of patent protection for various blockbuster drugs. Merck sought to bolster its R&D pipeline by doubling its number of late-stage development compounds from 9 to 18. On March 9, 2009, Merck & Co. announced a definitive merger agreement to merge with Schering-Plough for approximately $41 billion. The deal combined Merck’s strengths with Schering-Plough’s leading franchises in areas like respiratory, oncology, and women’s health. It consolidated their cholesterol-lowering franchises: the two companies were already joint partners for the drugs Zetia (ezetimibe) and Vytorin (ezetimibe + simvastatin combo pill), and the merger allowed Merck to simplify and fully control this portfolio. The merger with Schering-Plough officially closed on November 3, 2009, transforming Merck into the world’s second-largest prescription drug maker at the time. Of course, Merck also in-housed Schering-Plough’s established hepatitis C franchise, which included its interferon-based products. This acquisition provided Merck with the infrastructure and expertise to support the development and launch of a new class of Hepatitis C medicine that dramatically shifted the treatment paradigm.
Wrench in the Works
In the 2000s, the standard of care for treating chronic Hepatitis C was a grueling endurance test. Patients faced up to 48 weeks of peginterferon and ribavirin, a combination that induced severe fatigue, flu-like symptoms, and depression. For many, the virus persisted, mocking the effort. The medical community needed something that could target the virus more precisely. But how?
Back in 1989, when Michael Houghton and his team at Chiron sequenced the Hepatitis C virus, they revealed its infernal machinery. They found that Hepatitis C had a positive-strand RNA genome that was approximately 9.6 kilobases long. Its 5’ Untranslated Region (5’ UTR) contained a highly structured internal ribosome entry site (IRES) that directed the host’s cellular machinery to initiate protein translation in a cap-independent manner. This was the most critical RNA domain for translation. It adopted a complex tertiary structure that bound directly to the 40S ribosomal subunit, bypassing the need for canonical eukaryotic initiation factors and starting to making protein right away. The virus’ single large open reading frame (ORF) encoded a large polyprotein precursor of roughly 3,000 amino acids in length. This polyprotein was processed by both host and viral proteases into at least 10 functional proteins (see below):
Structural Proteins: These were found at the N-terminus and included the Core protein (forming the viral nucleocapsid), as well as the envelope glycoproteins E1 and E2.
Non-Structural (NS) Proteins: These constituted the enzymatic machinery for viral replication and included NS2, NS3 (protease and helicase), NS4A (protease cofactor), NS4B, NS5A (a multi-functional protein involved in replication and assembly), and NS5B (the RNA-dependent RNA polymerase).
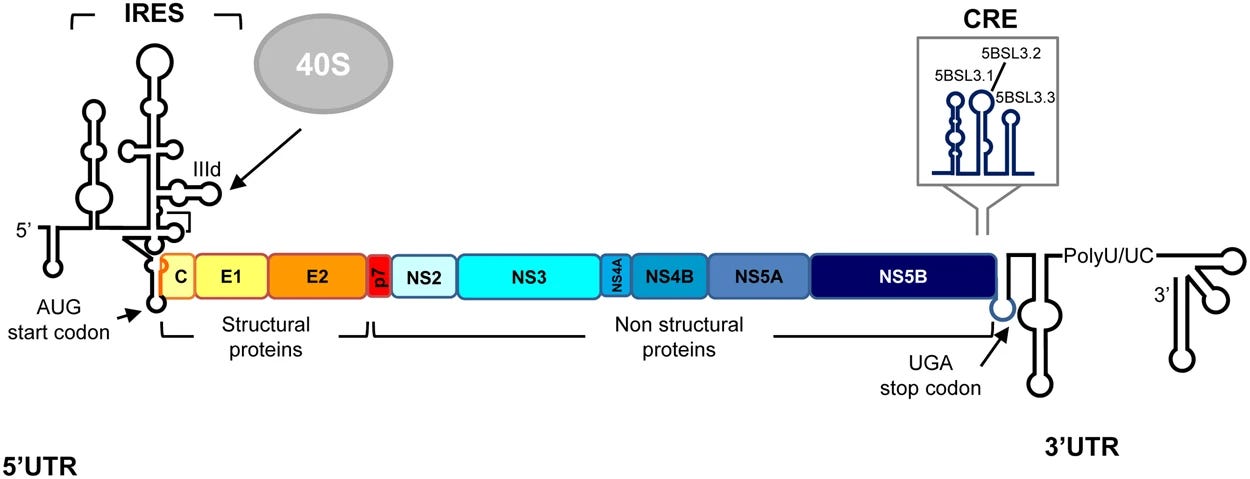
Specific stem-loop structures called the Cis-Acting Replication Elements (CREs) located within the coding regions (such as in the NS5B gene) and the UTRs were essential for the initiation of viral RNA synthesis. These structures acted as beacons for the viral RNA-dependent RNA polymerase (NS5B). Finally, the 3’ Untranslated Region (3’ UTR) was essential for viral replication and consisted of a variable region, a poly-U/UC tract, and a highly conserved 3’ X-tail, which formed the complex secondary RNA structures necessary for polymerase recognition. Out of all of these structures and substructures, scientists at Merck zeroed in on the NS3/4A protease for two reasons:
Essentiality for Viral Replication: As I mentioned before, the Hepatitis C genome produces a single, long polyprotein chain that must be cut into individual functional proteins to create new viral particles. Without the activity of the NS3 protease to perform these cuts, the virus cannot replicate. By blocking this molecular scissor, scientists could effectively halt the viral production line.
Druggable Binding Site: A major scientific discovery was that the NS3 protease is largely inactive on its own. It requires a viral protein called NS4A to act as a cofactor, which helps fold the protease into its active shape and anchor it to the cellular membrane where replication occurs. This specific NS3/4A complex provided a binding pocket for rational drug design that was different from any human proteases, theoretically allowing for a drug that would kill the virus while minimizing harm to the host’s normal cellular functions.
Once researchers solved the X-ray crystal structure of the NS3/4A complex, they could see the exact shape of the active site, a shallow groove where the viral polyprotein would normally fit. This allowed scientists to use structure-based drug design to create small molecules that mimicked the natural shape of the NS4A binding site (see graphic below). These molecules were designed to fit into that groove and act as a permanent, immovable plug, preventing the protease from cutting its natural substrate.
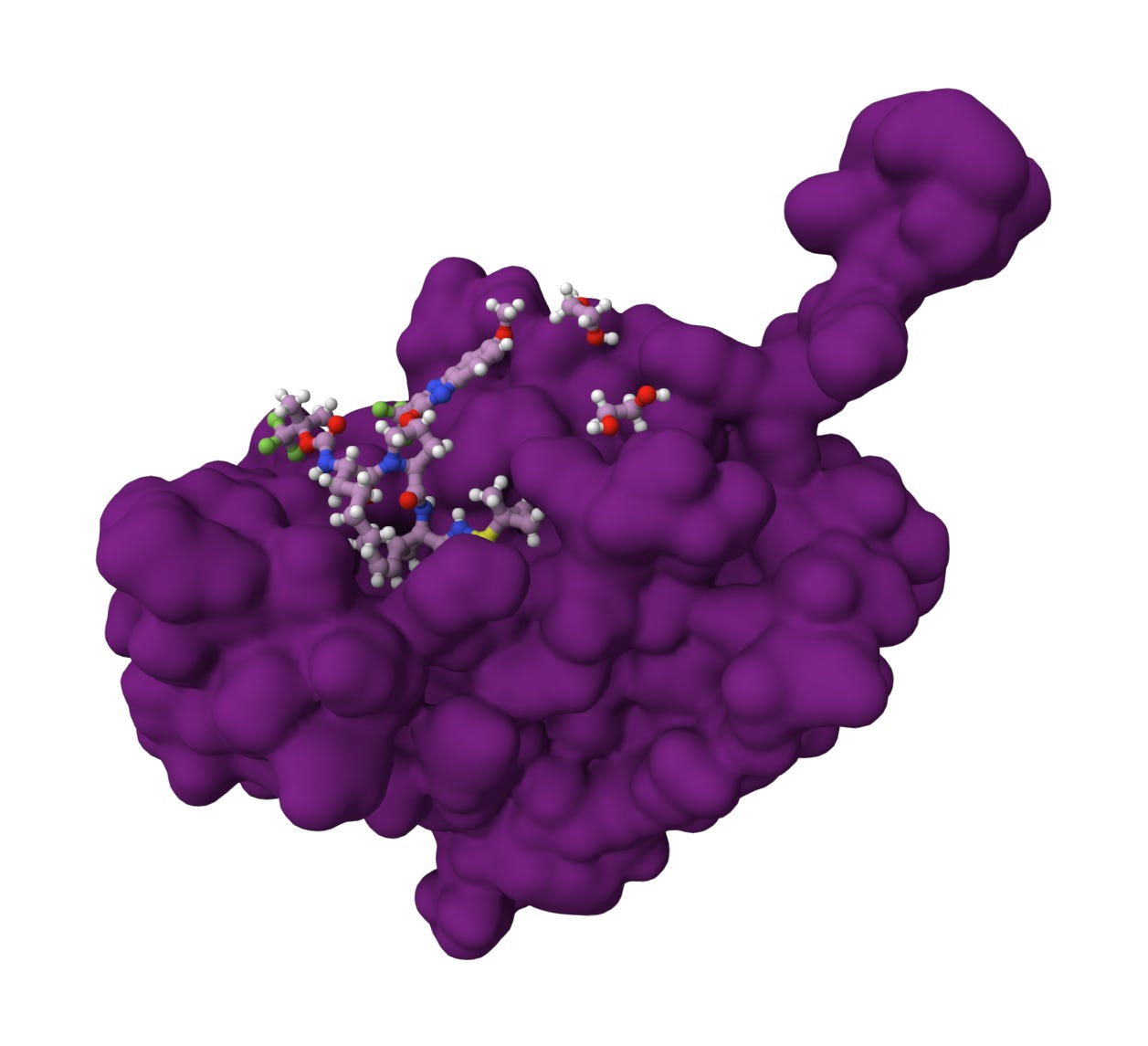
Originally initiated by Schering-Plough, Merck utilized a rigorous structure-based drug design process to develop Victrelis (boceprevir), the first direct-acting antiviral for Hepatitis C. Their starting point was an undecapeptide (a chain of 11 amino acids) with a high molecular weight of 1,265 Da, which readily occupied the NS4A binding site but was too bulky to use as an oral drug. Using X-ray crystallography to visualize the enzyme-inhibitor complex, researchers trimmed the peptide down to a tripeptide (about 500 Da). The resulting molecule was then fashioned as an α-ketoamide inhibitor. This electrophilic group acts as a trap for the serine-139 residue in the NS3 protease’s active site. When the inhibitor binds, it forms a reversible covalent bond, creating a tetrahedral intermediate that mimics the natural transition state of the protein-cleavage process. Next, medicinal chemists narrowed their focus into the bicyclic [3.1.0]proline moiety (the “P2” fragment). This specific structure served as a superior surrogate for the natural amino acid leucine, improving the drug’s binding potency by 1,000-fold.
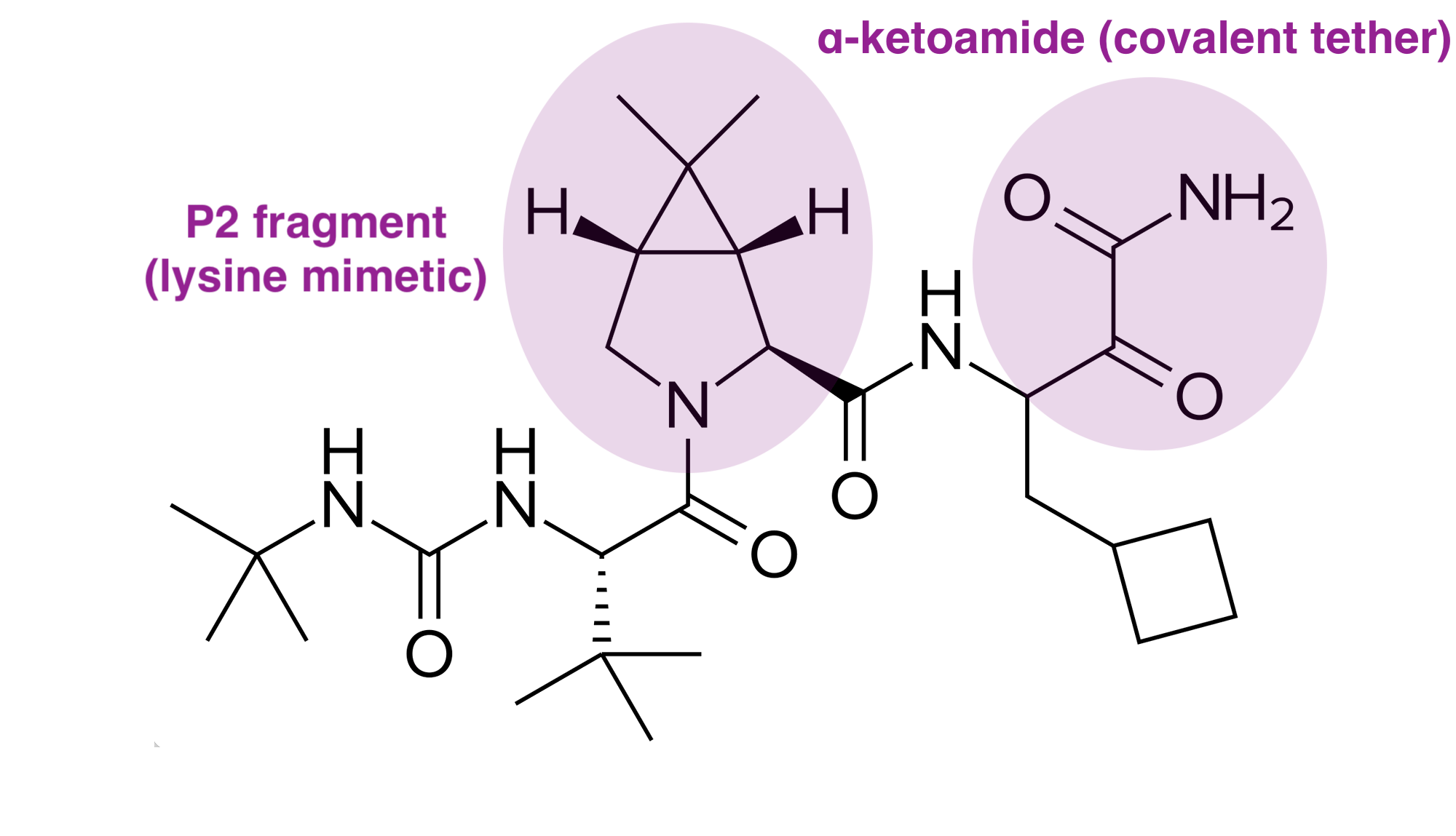
The design team faced significant hurdles in balancing potency with “drug-likeness” (pharmacokinetics). Early potent analogs were often charged (e.g., containing carboxylic acids), which prevented them from crossing cell membranes. Merck scientists substituted these groups with neutral alternatives, such as a dimethylamide, to enhance cell penetration. By systematically removing unnecessary residues at the “prime side” (the part of the inhibitor that extends out of the binding site) and identifying that non-substituted primary ketoamides maintained high potency, they successfully reduced the drug’s molecular weight into the range required for oral bioavailability.
This iterative, structure-guided approach allowed researchers to evolve a bulky, weakly active peptide into boceprevir, a potent, selective, and orally active inhibitor. By plugging the NS3 protease active site, the drug prevented the virus from cleaving its own polyprotein, thereby halting viral replication. This success served as a landmark proof-of-concept for the entire field of direct-acting antiviral (DAA) therapy. On May 13, 2011, that long-awaited shift finally arrived when the FDA approved Victrelis (boceprevir), supported by data from two pivotal, Phase 3 randomized, double-blind, placebo-controlled clinical trials:
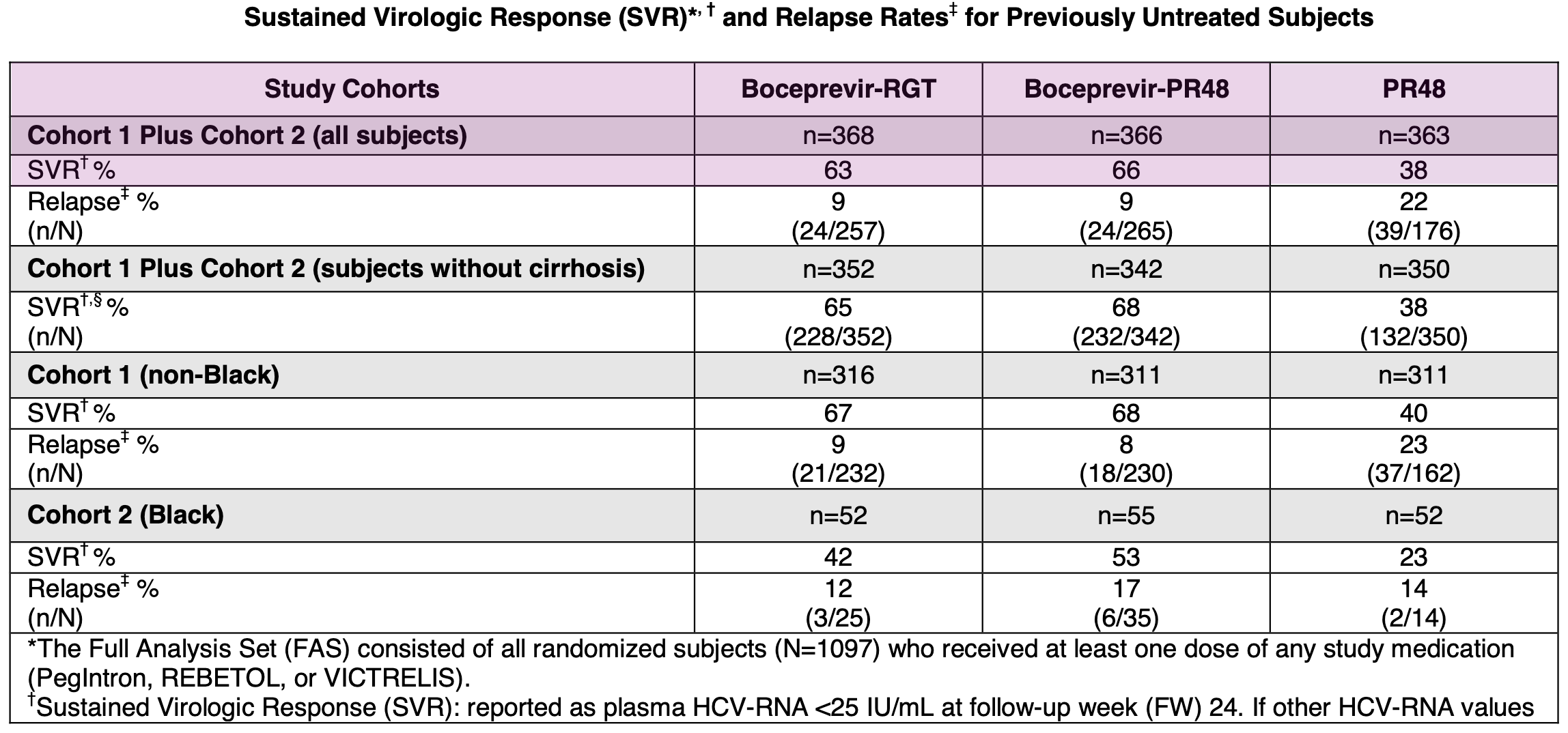
SPRINT-2 (Treatment-Naïve Patients): This trial enrolled 1,097 patients who had never previously been treated for Hepatitis C. Patients receiving boceprevir in combination with SOC achieved SVR rates of approximately 63-66%, compared to 38% for those receiving standard-of-care alone.
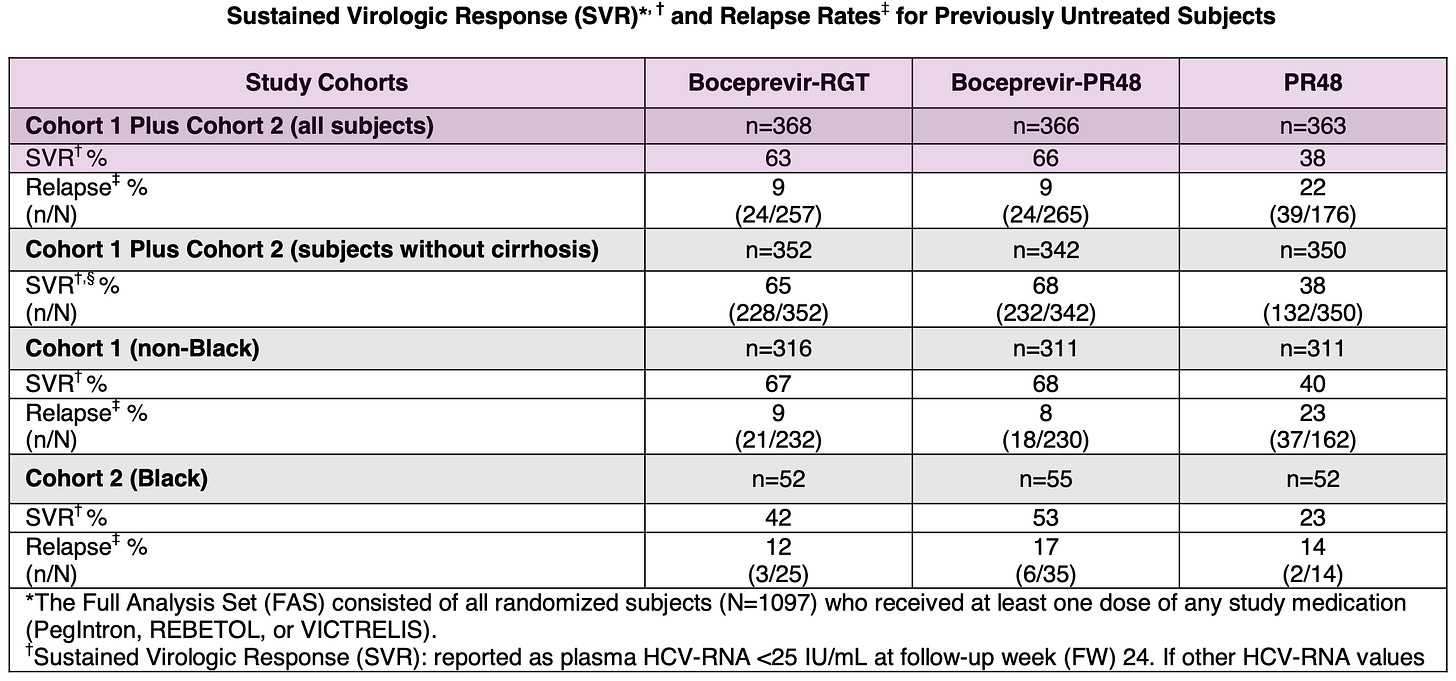
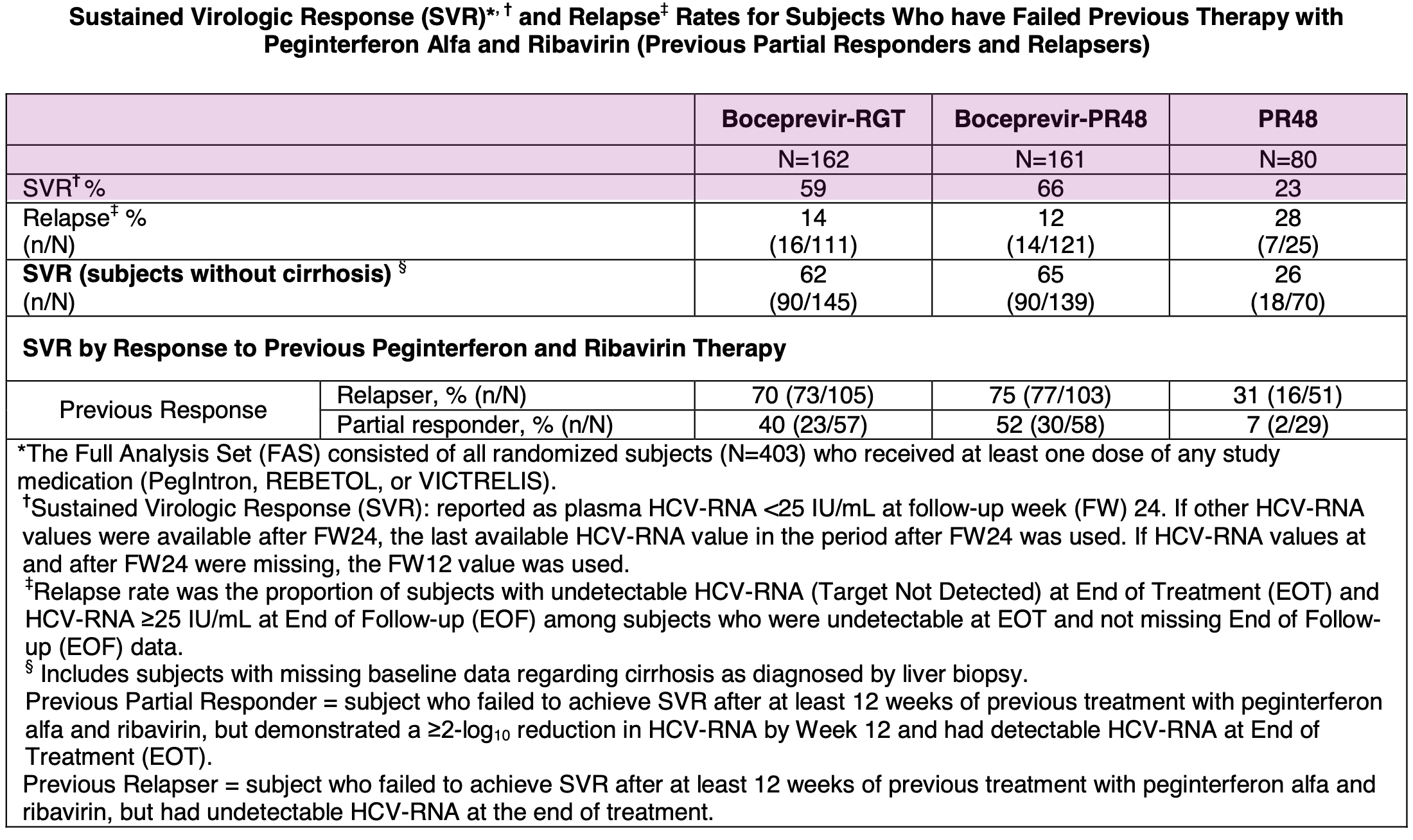
Results from Phase 3 SPRINT-2 trial in treatment-naïve Hepatitis C patients; Source: Victrelis label, Table 10 RESPOND-2 (Treatment-Experienced Patients): This trial enrolled 403 patients whose prior therapy with pegylated interferon and ribavirin had failed, including relapsers and previous non-responders. The addition of boceprevir similarly resulted in a significantly higher SVR rate of 59-66% compared to 23% in patients who received only the standard-of-care.
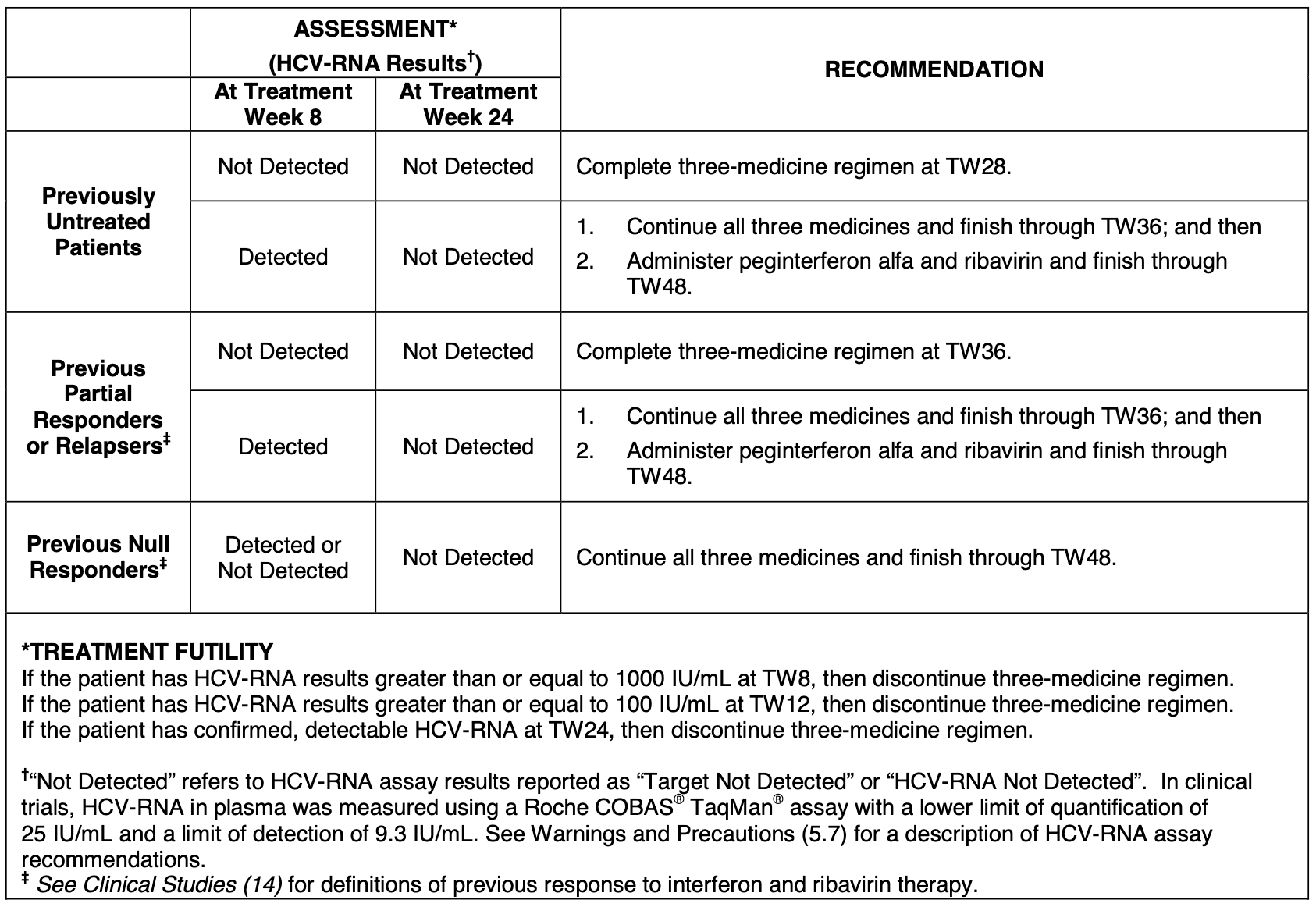
Since Victrelis markedly increased cure rates, it allowed physicians to adopt response-guided therapy. Patients who showed a strong, early virologic response could potentially shorten their total treatment duration to 28 weeks, sparing them months of the severe, flu-like side effects associated with the longer 48-week interferon-based course. A testing algorithm was included within the FDA label itself (see below), providing crucial guidance to the physician community.
Protease Pile-on
Merck’s wasn’t the only company with a crack team of medicinal chemists working on this problem. Vertex Pharmaceuticals was founded in 1989 by Joshua Boger, who had previously spent a decade at Merck as the Senior Director of Basic Chemistry and headed the Departments of Biophysical Chemistry and Medicinal Chemistry of Immunology & Inflammation. During his time there, he became a pioneer in the application of computer modeling to drug design, effectively organizing one of the first major efforts in what is now known as structure-based drug design.
In fact, Vertex was established specifically to be the first pharmaceutical company built entirely around the strategy of structure-based drug design. While other companies utilized this approach, Vertex made it the core, defining philosophy of its entire R&D operation, relying on computer modeling to visualize and manipulate the 3D structures of proteins and enzymes to design custom inhibitors. Vertex’s approach stood in contrast to the prevailing combinatorial chemistry or trial-and-error methods common at the time. Their work, including early efforts to target HIV protease, helped demonstrate the power of rational, structure-guided discovery. The story of Vertex’s early years, including its intense focus on these scientific pioneers and their rational design methods, was famously chronicled in Barry Werth’s 1994 book, The Billion-Dollar Molecule, which I highly recommend.
However, the Vertex of 2011 was not the same as the Vertex of 2026. Back then, Vertex was know as “The Hepatitis C” company rather than its modern moniker as “The Cystic Fibrosis” company. In 2011, Vertex was only in the final stages of its pivotal Phase 3 clinical program for CFTR corrector ivacaftor. They had just announced positive Phase 3 STRIVE results (in February 2011) showing significant improvements in lung function for patients with a specific cystic fibrosis mutation. They were preparing to submit regulatory applications for approval in the U.S. and Europe by the second half of 2011, which led to the drug’s approval (as Kalydeco) in early 2012. In parallel to Merck’s efforts, Vertex and collaborators Mitsubishi Tanabe Pharma Corporation & Janssen had put substantial weight behind their NS3/4A protease inhibitor Incivek (telaprevir), which was approved just 10 days after Victrelis on May 23, 2011 after their Phase 3 ADVANCE trial in treatment-naïve patients showed a 79% SVR rate compared to 46% in patients given standard of care. Incivek for Hepatitis C was, in fact, Vertex’s very first approved medicine.

While Merck’s Victrelis and Vertex’s Incivek were both peptidomimetic inhibitors designed to block the NS3 protease active site, their core chemical compositions featured distinct structural choices. Like Victrelis, Incivek utilized an α-ketoamide warhead, but its scaffold was distinctly optimized to fit the protease’s binding groove differently. Vertex’s approach prioritized a structure that offered high potency but, ultimately, presented unique challenges regarding formulation and administration. The most significant differences emerged during clinical use. Post-market reports identified serious and sometimes fatal skin reactions, which led to the FDA adding a black box warning to its label in 2012.

A year later, Vertex’s partner Janssen arrived on the scene with a follow-up compound. This time, Janssen partnered with Medivir to design a second generation NS3/4A protease inhibitor, characterized by structural and pharmacological improvements. First generation inhibitors utilized a linear, α-ketoamide chemical structure, forming a reversible covalent bond with the catalytic serine residue in the Hepatitis C NS3/4A protease. Janssen’s new molecule Olysio (simeprevir) was a macrocyclic compound. This cyclic structure was engineered to lock the molecule into a shape that fits more precisely and securely into the protease’s binding site, allowing for higher binding affinity and greater specificity. Clinically, this translated to improved potency and a more favorable side-effect profile compared to the first-generation drugs. On the convenience front, simeprevir in combination with peginterferon and ribavirin reduced patients’ pill burden to three pills a day compared to four pills a day for Incivek and five pills a day for Victrelis.
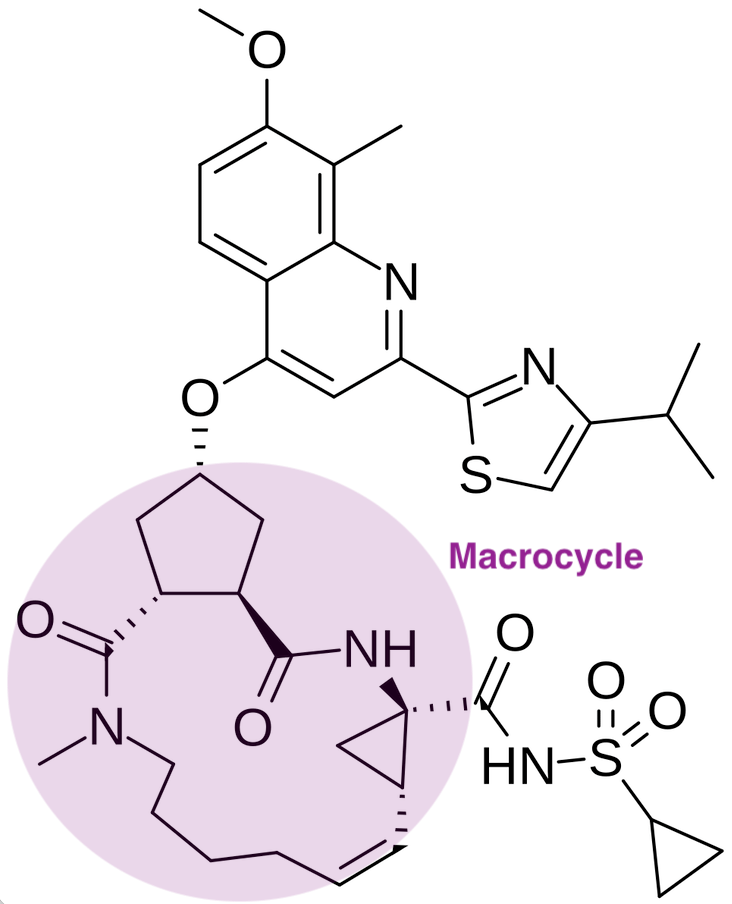
The approval of Olysio (simeprevir) on November 22, 2013 was primarily supported by three pivotal Phase 3 clinical trials:
QUEST-1 and QUEST-2 (Treatment-Naïve Patients): A pooled analysis of these studies showed that 80% of treatment-naïve patients receiving Olysio achieved a sustained virologic response 12 weeks after the end of treatment (SVR12), compared to 50% in the placebo groups.
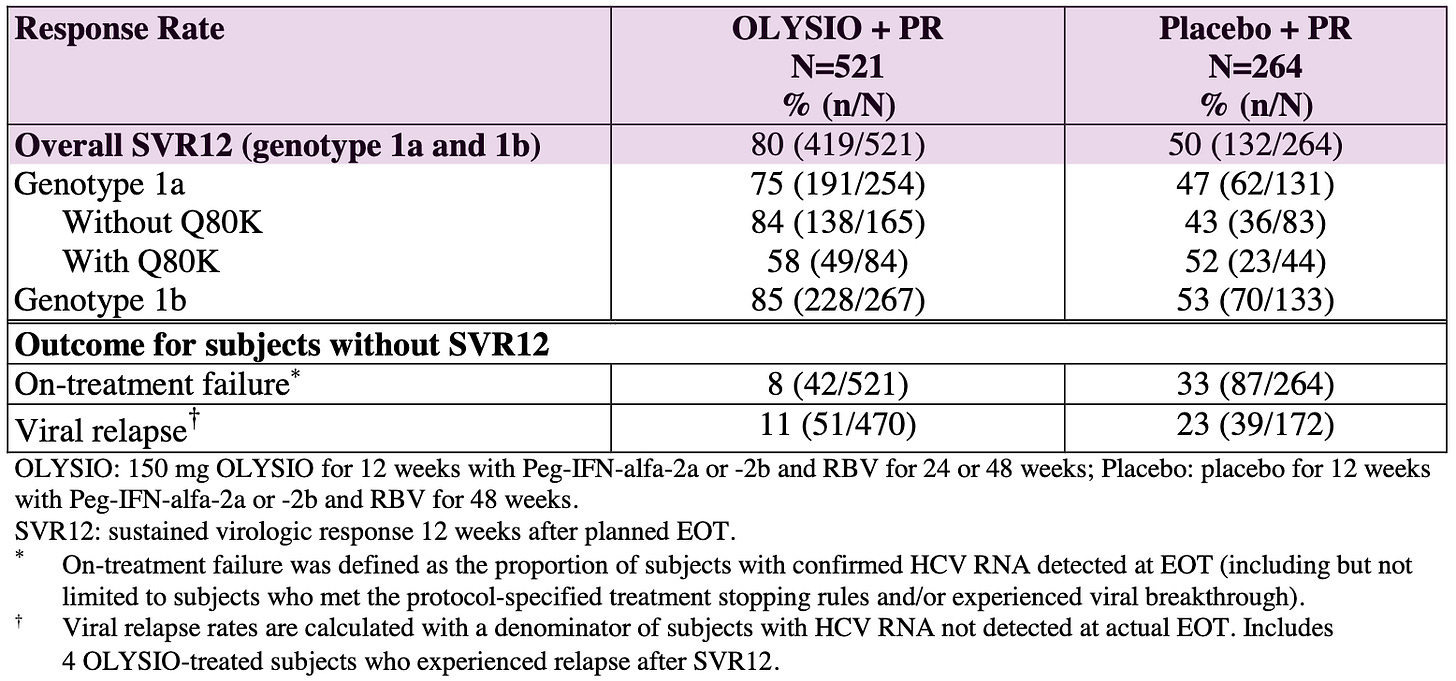
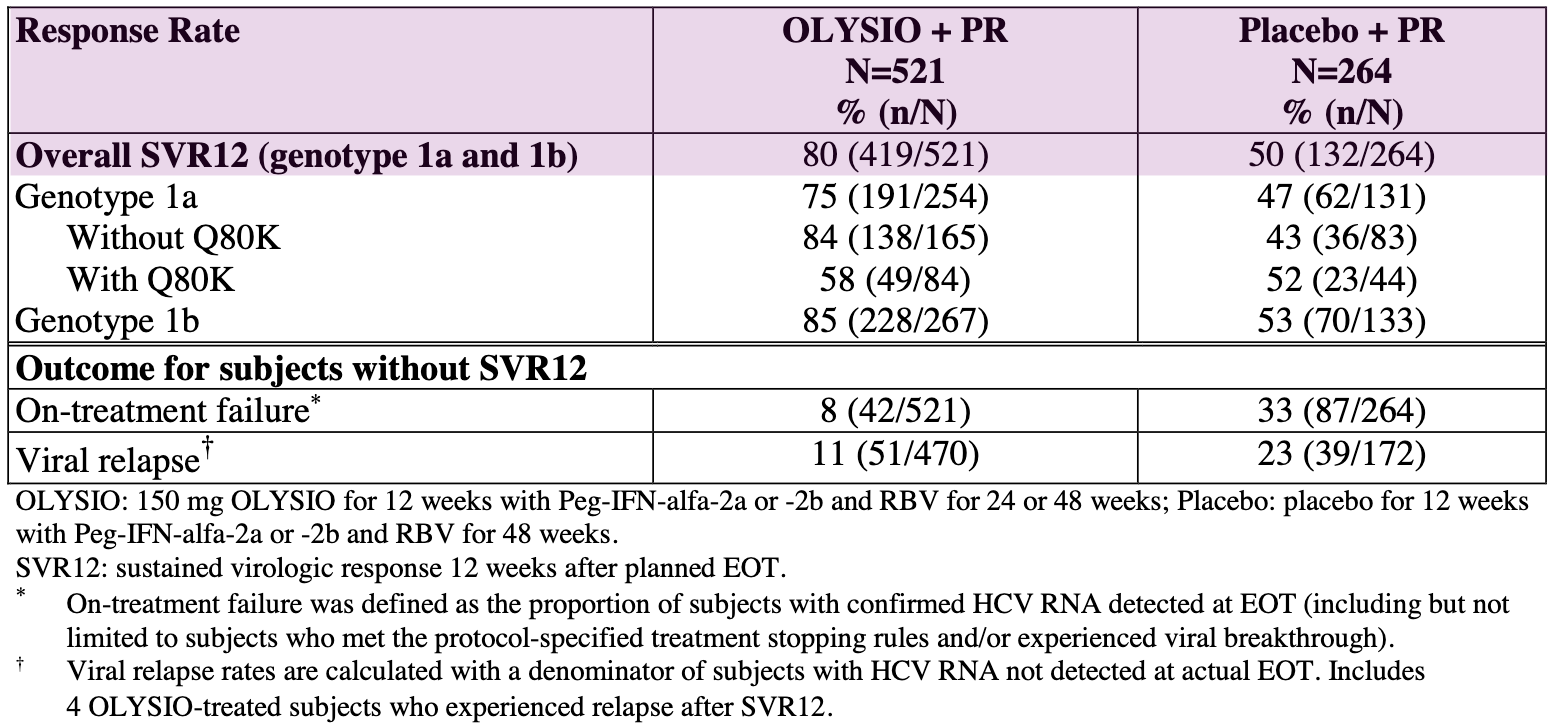
Virologic Outcomes in Treatment-Naïve Adult Subjects with HCV Genotype 1 Infection (Pooled Data QUEST 1 and QUEST 2 Trials); Source: Olysio label, Table 17 PROMISE (Treatment-Experienced Patients): This trial focused on patients who had relapsed after prior interferon-based treatment. In this study, 79% of patients in the Olysio group achieved SVR12, compared to 37% in the placebo group.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
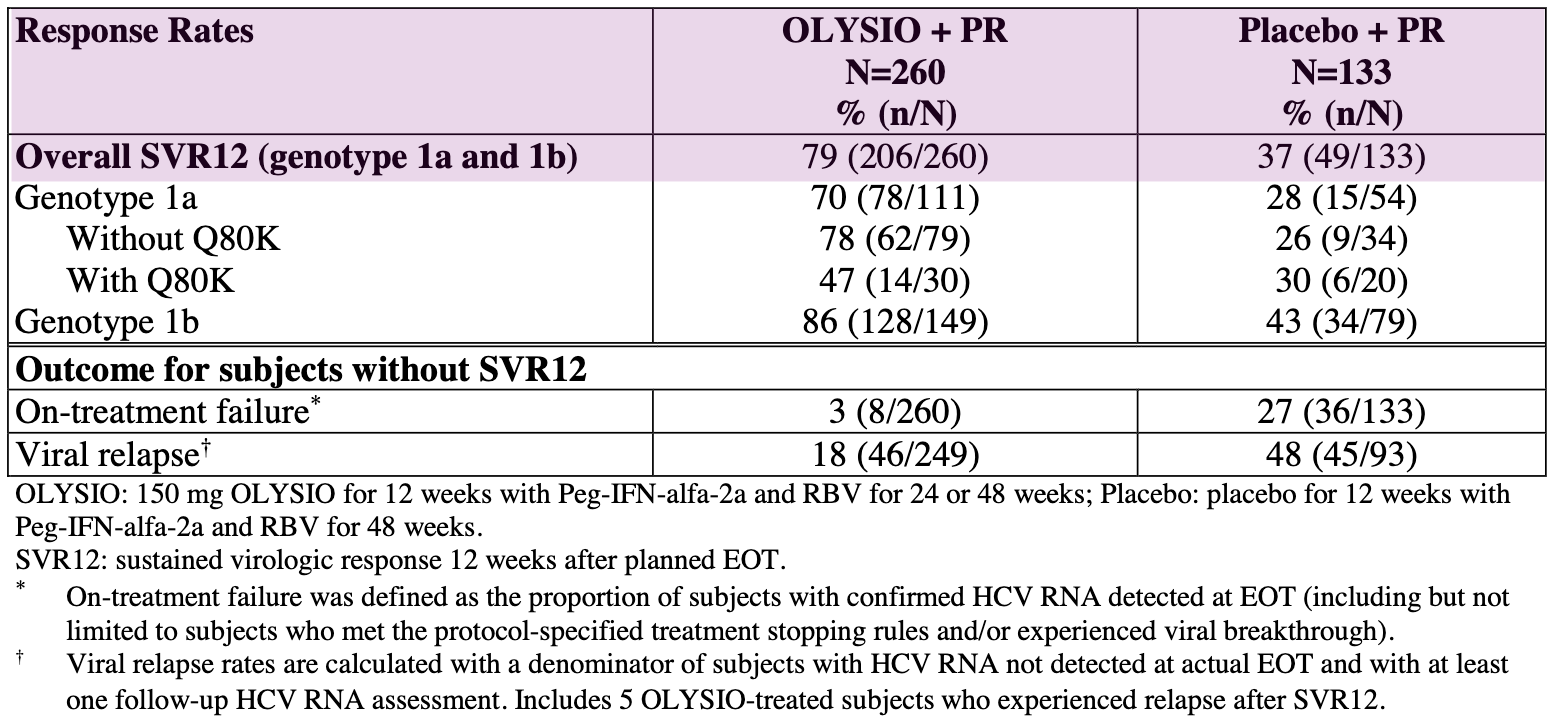
However, it wasn’t all smooth sailing for Janssen. During Olysio’s clinical development and subsequent review, it was discovered that its efficacy was significantly lower in patients infected with the HCV genotype 1a virus that contained the Q80K polymorphism, a naturally occurring variation in the virus’s protease enzyme. As a result, the FDA label included a recommendation to screen for this mutation and consider alternative therapies for these patients. Olysio’s liability and Incivek skin toxicity reminds us of the unpredictable nature of small molecule drug design: a single out-of-place atom can dramatically alter a drug’s pharmacokinetics and pharmacodynamics in ways that are difficult to detect until its tested in a 100+ patient Phase 3 trial.
Sun Sets on the Protease Era
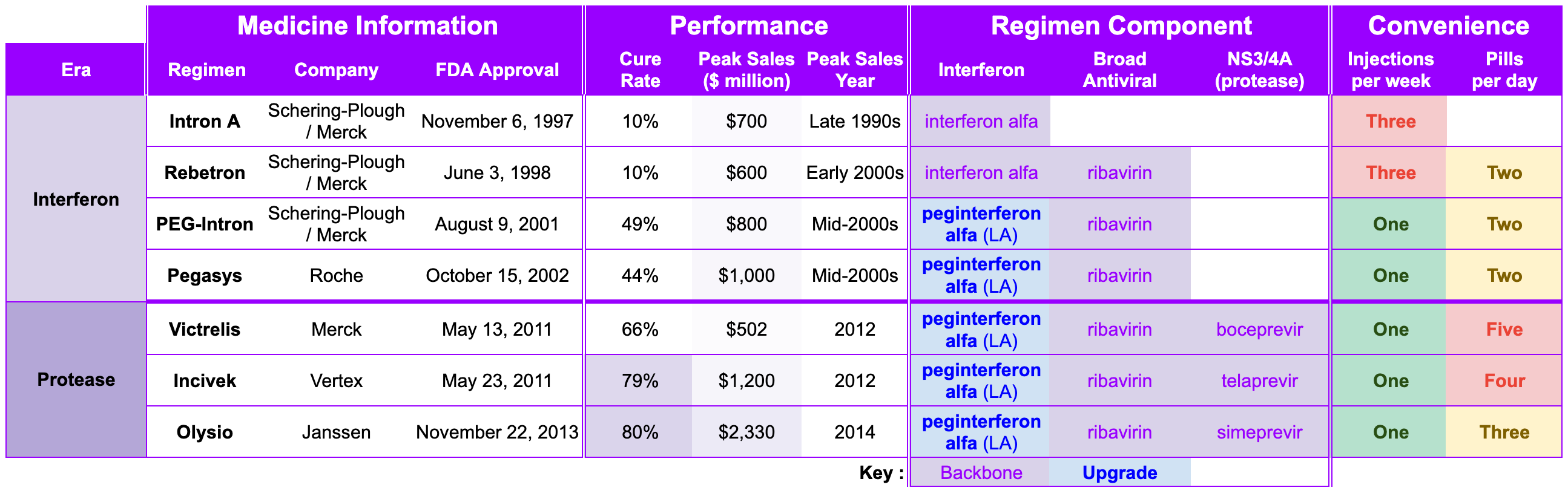
The Interferon Era and Protease Era represent the first two legs of the race for the Hepatitis C cure. Researchers at Chiron mapped the virus’ genome and developed a biomarker-based diagnostic, revealing the enormous scope of Hepatitis C’s scourge. Schering-Plough figured out how to manufacture interferon at scale, pioneering the first FDA-approved treatment for Hepatitis C. Schering-Plough and Roche introduced a long-acting peginterferon, which reduced the injection burden from three times weekly to once weekly. After Schering-Plough merged with Merck, the team hosted a second act and pioneered NS3/4A protease inhibitors with Vertex close behind. By the end of 2013, Schering-Plough/Merck had a 16-year track record as the juggernaut in Hepatitis C. Two years later, Janssen lowered the overall pill burden from five a day to three a day. All told, Hepatitis C went from a public health epidemic to an 80% cure rate.
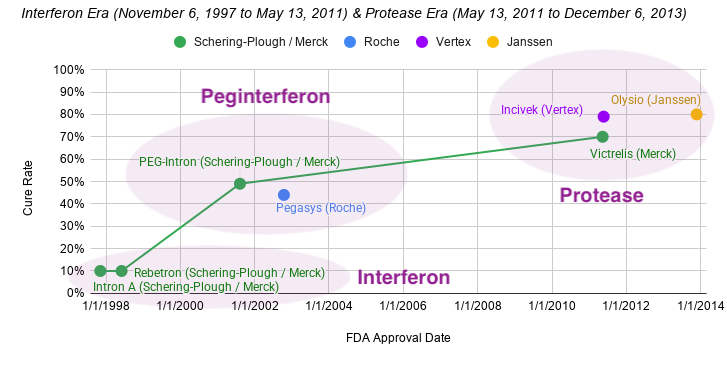
In a vacuum, this progress is astounding, but the story doesn’t end here. It’s only with the clarity of hindsight that we can talk about the weaknesses of the standard of care regimen for Hepatitis C circa 2013. NS3/4A protease inhibitors could not be used alone. They required co-administration with pegylated interferon and ribavirin, which were notorious for debilitating side effects like severe fatigue, depression, flu-like symptoms, and anemia. These regimens required complex dosing schedules (e.g., every 8 hours) and specific dietary protocols (Victrelis with a light snack and Incivek with a high-fat meal) to ensure proper absorption. Since they had a low barrier to resistance, any lapse in adherence could quickly render the virus resistant not only to the drug being taken but potentially to other protease inhibitors in the same class.
Beyond the specific issues of the first-generation drugs, the protease inhibitor class as a whole faced persistent clinical challenges. Early protease inhibitors were contraindicated or carried strong warnings for patients with moderate to severe liver impairment (Child-Pugh B or C cirrhosis). In these cases, the drugs could cause further liver dysfunction, failure, and death, likely due to altered hepatic metabolism. Protease inhibitors were also typically potent inhibitors or substrates of cytochrome P450 enzymes (particularly CYP3A). This made them prone to significant interactions with a wide array of other medications, necessitating careful clinical management when treating patients with comorbidities. Finally, the efficacy of certain protease inhibitors (such as Olysio) was compromised by pre-existing viral resistance mutations, most notably the Q80K polymorphism in HCV genotype 1a. This required mandatory baseline resistance testing, adding complexity and cost to the treatment process.
As the sun set on the Protease Era, Shering-Plough/Merck looked back on a 16-year track record of success and a blockbuster Hepatitis C franchise (see below). Roche, Vertex, and Janssen also constructed blockbuster franchises, racking up peak annual sales of $1 billion (mid-2000s), $1.2 billion (2012), and $2.3 billion (2014), respectively. High fives all around.
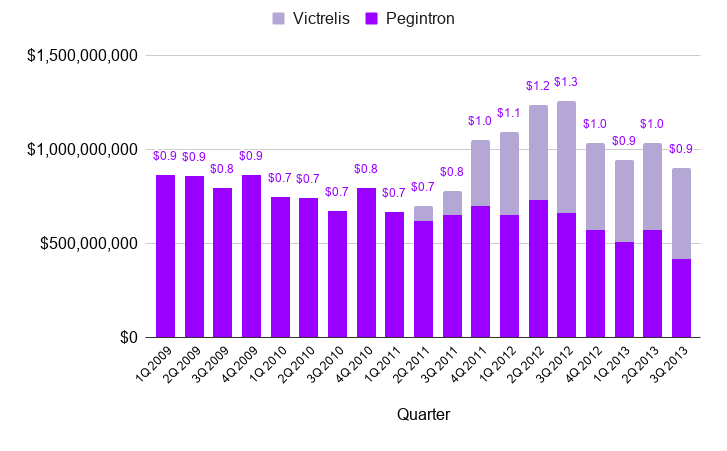
Hold a second. Something is missing.
Remember Gilead, the underdog of our story? They’re about to shift into overdrive. The Gold Rush has barely begun. The race for the cure is just getting started.
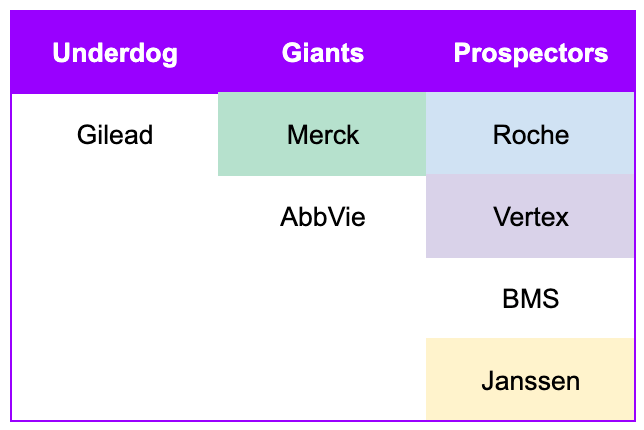
Stick around for Part 2, where we explore how Gilead bested big pharma in the race for a cure.
Subscribe so you don’t miss it!
If you enjoyed Part 1, don’t forget to like and share.
Love biotech? Check out Biotech Readout’s full library of content library, or navigate directly to a segment that interests you:
Frontiers in Medicine: Exploring the frontiers of our understanding and treatments for disease.
Medical History: Recovering forgotten relics in the history of medicine.
Acquisitions: Exploring the innovation behind acquired companies.
Weekly Readout: A digest of new clinical data from the past week.

To contact us, please send us an email at biotechreadout@gmail.com
Disclaimers
Investigational Status Disclaimer
The therapeutic candidates discussed in this newsletter are currently in clinical development and have not been approved for commercial sale by the U.S. Food and Drug Administration (FDA), the European Medicines Agency (EMA), or other global regulatory authorities. Their safety and efficacy have not been established. References to pipeline products and ongoing clinical trials involve significant risks and uncertainties. Statements regarding the potential safety, potency, or efficacy of investigational drugs reflect current hypotheses and are not a guarantee of future performance or regulatory clearance. The outcome of clinical trials is inherently unpredictable, and clinical results from earlier stages may not be predictive of results in later, larger-scale trials.
No Medical Advice Disclaimer
This newsletter is for informational and educational purposes only. The content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or another qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read in this publication.
No Patient-Provider Relationship Disclaimer
The information provided in this newsletter is for educational and analytical purposes only. Receipt of this information, or any interaction with this content, does not create a physician-patient, pharmacist-patient, or any other professional-provider relationship between you and the authors or publishers. This newsletter should not be used as a substitute for a personal consultation with a qualified healthcare professional.
Forward-Looking Statements Disclaimer
This newsletter contains “forward-looking statements” regarding future events, including clinical trial timing, regulatory milestones, and projected market performance. These statements are based on current expectations and assumptions that are subject to significant risks and uncertainties. Actual results may differ materially from those expressed or implied. We undertake no obligation to update these statements as a result of new information or future developments.
Third-Party Links & Content Disclaimer
This newsletter contains links to third-party websites, including clinical trial registries and corporate presentations. Biotech Readout does not endorse, guarantee, or assume responsibility for the accuracy or reliability of any information offered by third-party providers.
Errors and Omissions Disclaimer
While we strive for technical accuracy, the information in this newsletter is provided on an “as is” basis with no guarantees of completeness, accuracy, or timeliness. Biotech Readout assumes no liability for any errors or omissions in the content of this publication.
Non-Endorsement Disclaimer
Any reference to specific commercial products, processes, or services by trade name, trademark, or manufacturer does not constitute or imply an endorsement or recommendation by the author. All trademarks are the property of their respective owners.
No Investment Advice Disclaimer
This newsletter is for informational purposes only and does not constitute financial, investment, or legal advice. The author is not a registered investment advisor. You should consult with a professional financial advisor before making any investment decisions. The biotechnology sector is highly volatile; past performance is not indicative of future results.
Conflict of Interest Disclaimer
The author of this newsletter maintains a position of independence. At the time of publication, the author holds no direct financial interest, equity, or options in any of the companies mentioned in this report. No compensation has been received from any third party to feature or analyze specific therapeutic candidates or corporate entities.