
GSK to Acquire Nuvalent
Bolting on a three-headed hydra of targeted lung cancer drugs

Disclaimer: This newsletter is for educational and informational purposes only and does not constitute medical, investment, or financial advice, nor does it establish a provider-patient relationship. Content may include forward-looking statements and discussions of investigational therapeutic candidates that are not FDA/EMA approved; their safety and efficacy remain unestablished and clinical outcomes are unpredictable. While we strive for accuracy, all information is provided as is without guarantees. This newsletter is independent, and the author holds no financial positions in the companies mentioned nor receives third-party compensation for this coverage. Please find a complete version of our disclaimers at the bottom of this article and on our About page.

Introduction
On June 9, 2026, GSK announced that it has entered an agreement to acquire Nuvalent, Inc. for $10.6 billion (GSK press release, GSK slide deck, Nuvalent press release). This acquisition centers on three targeted oncology assets:
Zidesamtinib (ROS1 inhibitor, PDUFA 9/18/2026)
Neladalkib (ALK inhibitor, PDUFA 11/27/2026)
NVL-330 (HER2 inhibitor, Phase 1 ongoing)
While most biotech buyouts focus on a single crown jewel, GSK has acquired a diversified “stable” of molecules that hits three mutations associated with non-small cell lung cancer (NSCLC). At first glance, a general industry analyst might look at the pie chart and question the value proposition: ALK+ (4%), ROS1+ (2%), and HER2 (2%) combined represent a seemingly small slice (~8%) of the total non-small cell lung cancer (NSCLC) market. Large-cap pharmaceutical companies traditionally target broad populations like KRAS mutations (25%) or the massive “Unknown/Other” chemotherapy and immunotherapy pools.
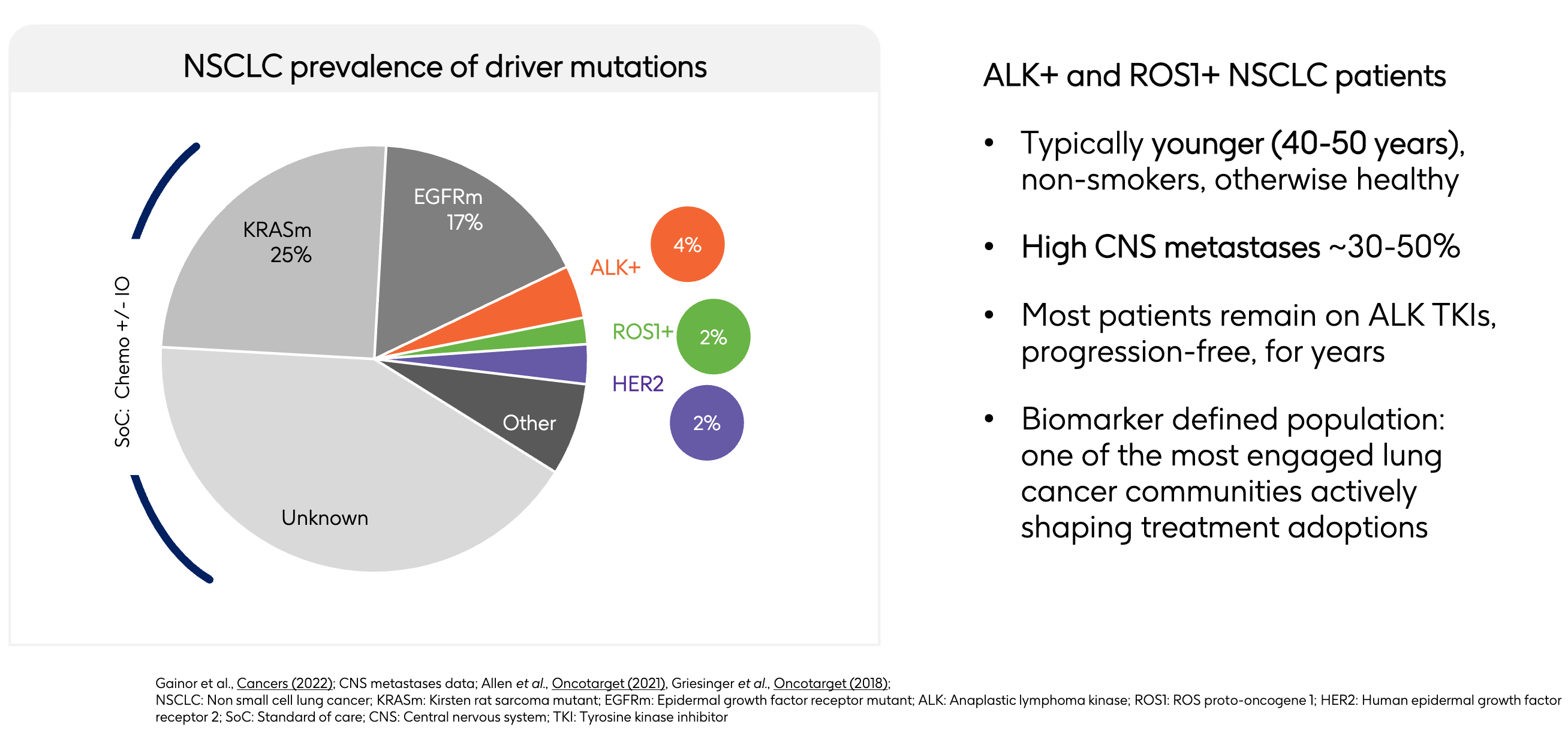
However, GSK’s expansion layout highlights the under-appreciated commercial value of this 8% slice. Patients with KRAS mutations or unknown drivers are subjected to standard chemotherapy combined with immuno-oncology (IO) regimens. These markets are highly crowded, hyper-competitive, and prone to rapid therapy switching due to inconsistent durability. By acquiring Nuvalent’s portfolio (neladalkib, zidesamtinib, and NVL-330), GSK aims to establish a strategic position across three distinct, biomarker-defined segments. In precision medicine, dominating three highly validated, 2% to 4% niches with potential best-in-class assets could yield more predictable, high-margin revenue than competing in a broad, fragmented market. GSK expects the acquisition “to be accretive to sales and core operating profit in 2027 and core EPS in 2029 inclusive of synergies and reprioritisation.”
Here, we dive into the M&A strategy that GSK highlighted in their presentation, the discovery & drug development generations for ALK & ROS1 inhibitors, and the corporate history of Nuvalent.
A Peek Into GSK’s M&A Strategy
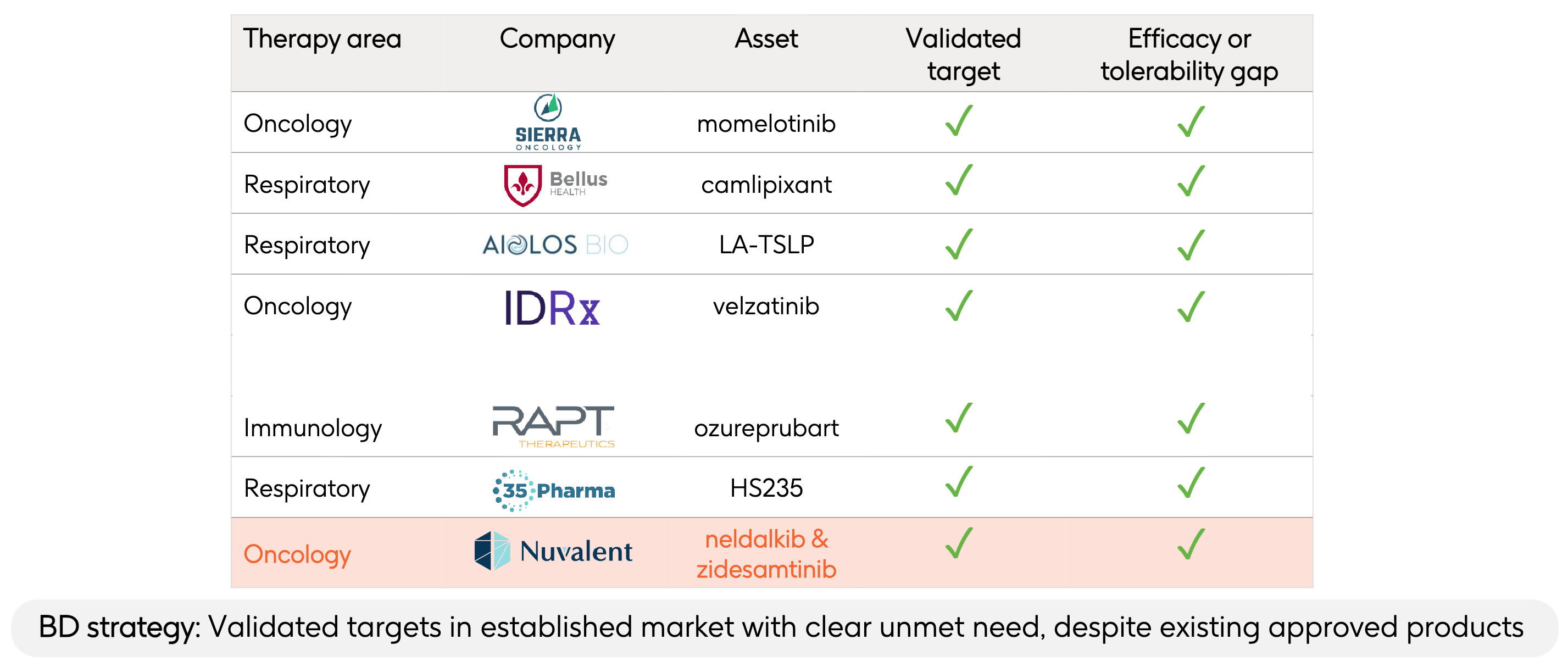
During GSK’s presentation regarding its $10.6 billion acquisition of Nuvalent, Chief Executive Officer Luke Miels emphasized that this transaction represents a direct continuation of their established Business Development (BD) framework. The slide above highlighted previous examples where GSK acquired companies that they deemed to have a “validated target” that fixed an “efficacy or tolerability gap”:
Sierra Oncology / Momelotinib (Oncology): The standard of care for myelofibrosis (like ruxolitinib) relies heavily on JAK1/JAK2 inhibition, which routinely suppresses bone marrow production and causes severe, treatment-limiting anemia and thrombocytopenia. Momelotinib uniquely inhibits ACVR1 (in addition to JAK1/JAK2), downregulating hepcidin to actually restore iron homeostasis and red blood cell production, fixing the anemia gap.
BELLUS Health / Camlipixant (Respiratory): Early-generation P2X3 antagonists developed for refractory chronic cough suffered from a major tolerability issue: they frequently blocked the P2X2 receptor subtype, causing severe taste disturbance (dysgeusia) or total taste loss, leading to high clinical discontinuation rates. Camlipixant was engineered as a highly selective P2X3 antagonist that spares P2X2, preserving normal taste sensation while maintaining potent cough-reflex suppression.
Aiolos Bio / LA-TSLP (Respiratory): Approved anti-TSLP antibodies for severe asthma (such as tezepelumab) require frequent subcutaneous injections every 4 weeks. This high treatment burden impacts long-term patient adherence and lifestyle flexibility. LA-TSLP is a long-acting monoclonal antibody designed with an extended half-life to enable every-6-month dosing, converting a frequent clinical intervention into a twice-a-year maintenance regimen.
IDRx / Velzatinib (Oncology): Existing tyrosine kinase inhibitors (TKIs) used for gastrointestinal stromal tumors (GIST), such as imatinib, inevitably succumb to resistance. Over 90% of progressing patients develop highly heterogeneous, secondary resistance mutations in exons 13, 14, 17, or 18 of the KIT gene, which standard TKIs fail to cover simultaneously. Velzatinib (IDRX-42) was specifically engineered to broadly inhibit the entire spectrum of clinically relevant primary and secondary KIT mutations, closing the evasion gap that causes tumor relapse.
RAPT Therapeutics / Ozureprubart (Immunology): Omalizumab (Xolair), the standard anti-IgE biologic for severe food allergies and chronic spontaneous urticaria (CSU), requires painful injections every 2 to 4 weeks. Additionally, a strict dosing ceiling leaves up to 25% of patients with high body weights or exceptionally high baseline IgE levels entirely ineligible for treatment. Ozureprubart is a half-life extended anti-IgE antibody engineered to allow dosing intervals of 8 to 12 weeks while boasting a broader therapeutic window capable of treating the high-weight/high-IgE patient populations previously locked out of therapy.
35Pharma / HS235 (Respiratory): First-generation activin signaling inhibitors used in pulmonary hypertension inadvertently cross-react with BMP9 and BMP10 ligands. This off-target binding causes structural vascular fragility, manifesting as a high risk of severe bleeding and telangiectasia (broken blood vessels), a major danger for patients concurrently taking blood thinners. HS235 features enhanced structural selectivity that deliberately spares BMP9 and BMP10, which could lower bleeding risks while adding distinct metabolic perks like fat-selective weight loss and improved insulin sensitivity.
Nuvalent also fits this paradigm. Early-generation ALK and ROS1 inhibitors for non-small cell lung cancer (NSCLC) fall short on three fronts:
Selectivity Gap: They cause severe neurological toxicities (dizziness, weight gain, cognitive changes) because they inadvertently inhibit the structurally similar TRK receptor family.
Efficacy Gap: When non-small cell lung cancer (NSCLC) is treated with early-generation inhibitors, the tumor cells adapt by developing secondary “solvent-front” or “gatekeeper” mutations. These mutations physically alter the shape of the binding pocket, blocking older drugs from entering.
ALK resistance: In ALK-positive lung cancer, successive lines of treatment (like crizotinib followed by alectinib or brigatinib) force the tumor to evolve highly complex single and compound ALK mutations. The most aggressive of these is the G1202R solvent-front mutation, as well as double mutations like 1151T/G1202R.
ROS1 resistance: The most notorious resistance adaptation in ROS1-positive NSCLC is the ROS1 G2032R solvent-front mutation. When this mutation occurs, standard therapies like crizotinib or entrectinib lose virtually all potency, leaving patients with no targeted options.
Delivery Gap: Up to 40% to 50% of patients with advanced ROS1- or ALK-positive NSCLC either present with or develop brain metastases during their treatment journey. Early-generation TKIs are poor substrates for crossing the blood-brain barrier, or they are actively pumped out of the brain by P-glycoprotein (P-gp) efflux pumps.
Both zidesamtinib and neladalkib were engineered to potentially address these limitations:
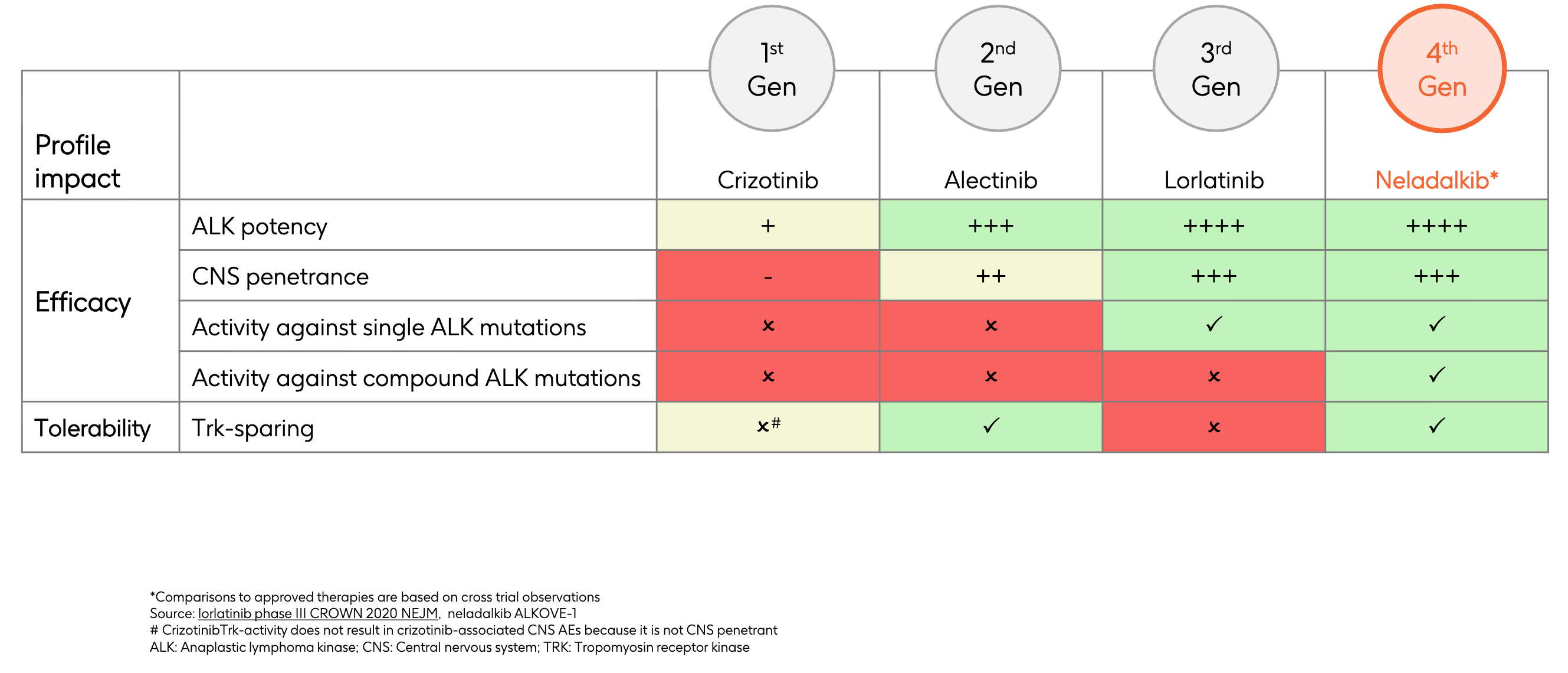
Selectivity Fix: Nuvalent designed both zidesamtinib and neladalkib to be highly TRK-sparing. They possess macrocyclic structures that are macro-selectively tuned to fit perfectly into the ATP-binding pockets of mutated ROS1 or ALK, but are structurally hindered from binding to TRK. This allows patients to stay on the highest therapeutic dose of the drug while minimizing debilitating cognitive or physical neurological side effects.
Efficacy Fix: Zidesamtinib was specifically engineered with a compact, rigid structure designed to avoid steric clash with the mutated, bulky arginine residue at position 2032. It binds tightly to both wild-type ROS1 and the mutated G2032R variant, shutting down the tumor’s primary resistance mechanism. Neladalkib is a potent, fourth-generation ALK inhibitor designed with a low molecular weight and optimized conformation that fits deeply into the ALK kinase domain. It maintains extreme potency across a broad spectrum of single and compound ALK mutations that render even third-generation inhibitors like lorlatinib ineffective.
Delivery Fix: Both molecules were built from the ground up to maximize cerebrospinal fluid (CSF) penetrance. They possess optimal lipophilicity and molecular weight profiles that bypass standard cellular efflux mechanisms. By achieving high, sustained concentrations inside the central nervous system, they are able to aggressively shrink existing brain metastases and prevent new intracranial lesions from forming, all while maintaining the TRK-sparing safety profile outlined above.
By targeting clinically validated oncology pathways, Nuvalent’s engineering has overcome the resistance and neurological tolerability gaps that limited earlier generations of TKIs. But, how did we find these “validated targets” in the first place?
(A)ttacking (L)ung (K)ancer
The ALK gene was first discovered in 1994 by researchers studying anaplastic large-cell lymphoma (ALCL), a rare form of non-Hodgkin lymphoma. They identified a recurring chromosomal translocation, designated as t(2;5)(p23;q35), which abnormally fused the ALK gene on chromosome 2 with the nucleophosmin (NPM) gene on chromosome 5. This historic finding established ALK‘s capability to act as an oncogene when structurally rearranged. However, because ALCL is relatively rare, the wider scientific community initially viewed ALK as a niche, disease-specific driver rather than a major target in solid tumors. For over a decade, it remained largely isolated to hematologic research.
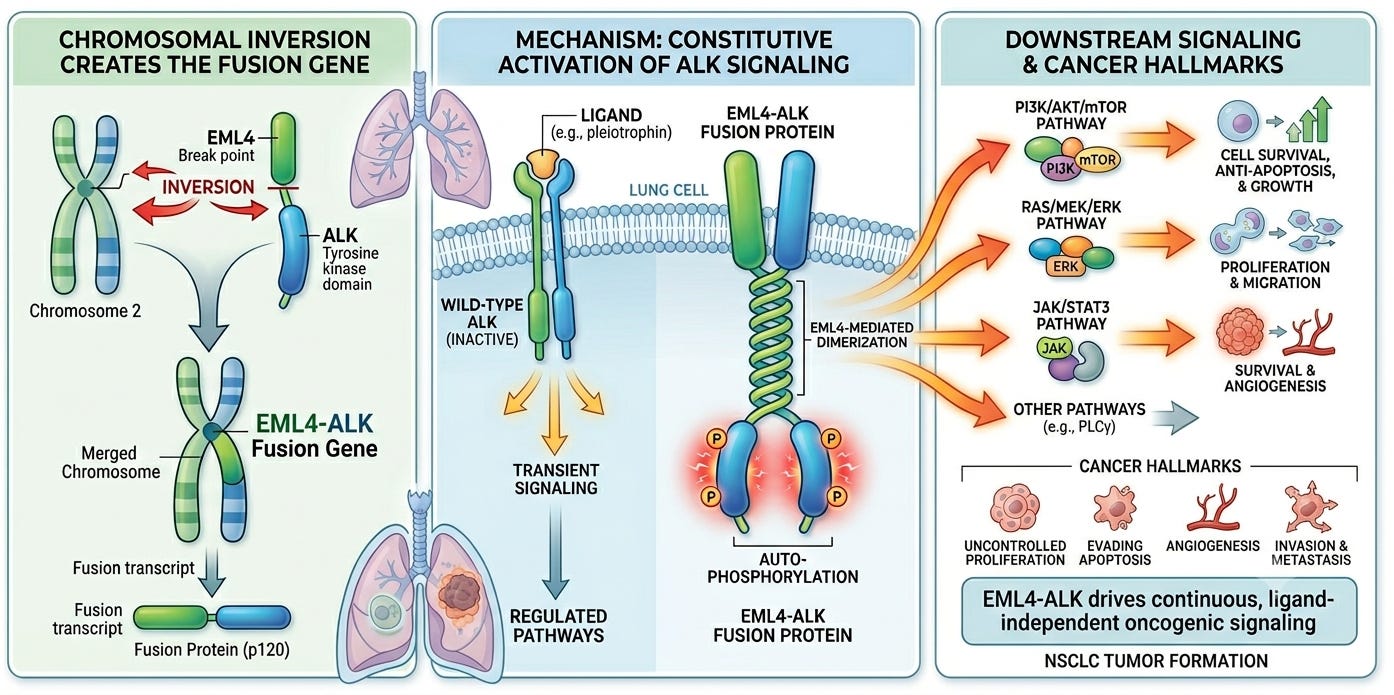
In 2007, a team of Japanese scientists including Dr. Manabu Soda and Dr. Hiroyuki Mano at the University of Tokyo revealed that there was more to the story. Emboldened by the recent discovery of EGFR mutations in non-small cell lung cancer (NSCLC), the team was systematically screening clinical specimens from lung adenocarcinoma patients to isolate novel transforming genes. Using a retroviral cDNA expression library generated from a patient’s tumor, they identified a crucial genetic error: a small, cryptic intrachromosomal inversion on the short arm of chromosome 2 (2p). This structural inversion disrupted and fused two adjacent genes:
The amino-terminal portion of Echinoderm Microtubule-Associated Protein-Like 4 (EML4)
The intracellular kinase domain of Anaplastic Lymphoma Kinase (ALK)
To prove this new EML4-ALK fusion was a true driver mutation, rather than an innocent bystander, the researchers forced its expression in mouse 3T3 fibroblasts. The cells rapidly formed transformed foci in culture and grew into large tumors when injected into nude mice, proving the fusion protein possessed potent, autonomous oncogenic competence. Simultaneously, a separate team utilized a global phosphoproteomic screen to independently identify identical ALK fusions across a broad screen of lung cancer cell lines.
Biochemically, the native, wild-type ALK receptor is a transmembrane tyrosine kinase that requires a specific extracellular ligand to activate. When the EML4-ALK fusion occurs, the extracellular ligand-binding domain and the transmembrane segment of ALK are entirely replaced by the amino-terminal coiled-coil domain of EML4. This specific structural region of EML4 has a natural, powerful tendency to pair up (dimerize). As a result, the attached ALK kinase domains are pulled together into a state of perpetual, ligand-independent dimerization and autophosphorylation. This unleashes a continuous, uncontrolled downstream signaling cascade through the RAS/MAPK, PI3K/AKT, and STAT3 pathways, forcing the lung cell into rapid, unhalted proliferation and cell survival.
As screening protocols expanded globally via fluorescence in situ hybridization (FISH) and RT-PCR assays, a distinct clinicopathologic profile emerged. While EGFR mutations were highly prevalent in East Asian women, the EML4-ALK fusion was found to be highly enriched in a completely distinct, distinct subset comprising roughly 3% to 7% of all NSCLC patients. The typical ALK-positive patient was significantly younger (often in their 40s or 50s, compared to a median lung cancer diagnosis age in the late 60s) and predominantly had a history of being a light smoker or having never smoked at all. This stark clinical demarcation transformed how the industry approached drug design. It suggested that advanced lung cancer could be a molecularly defined disease, paving a direct path for the immediate clinical deployment and rapid approval of targeted ALK inhibitors.
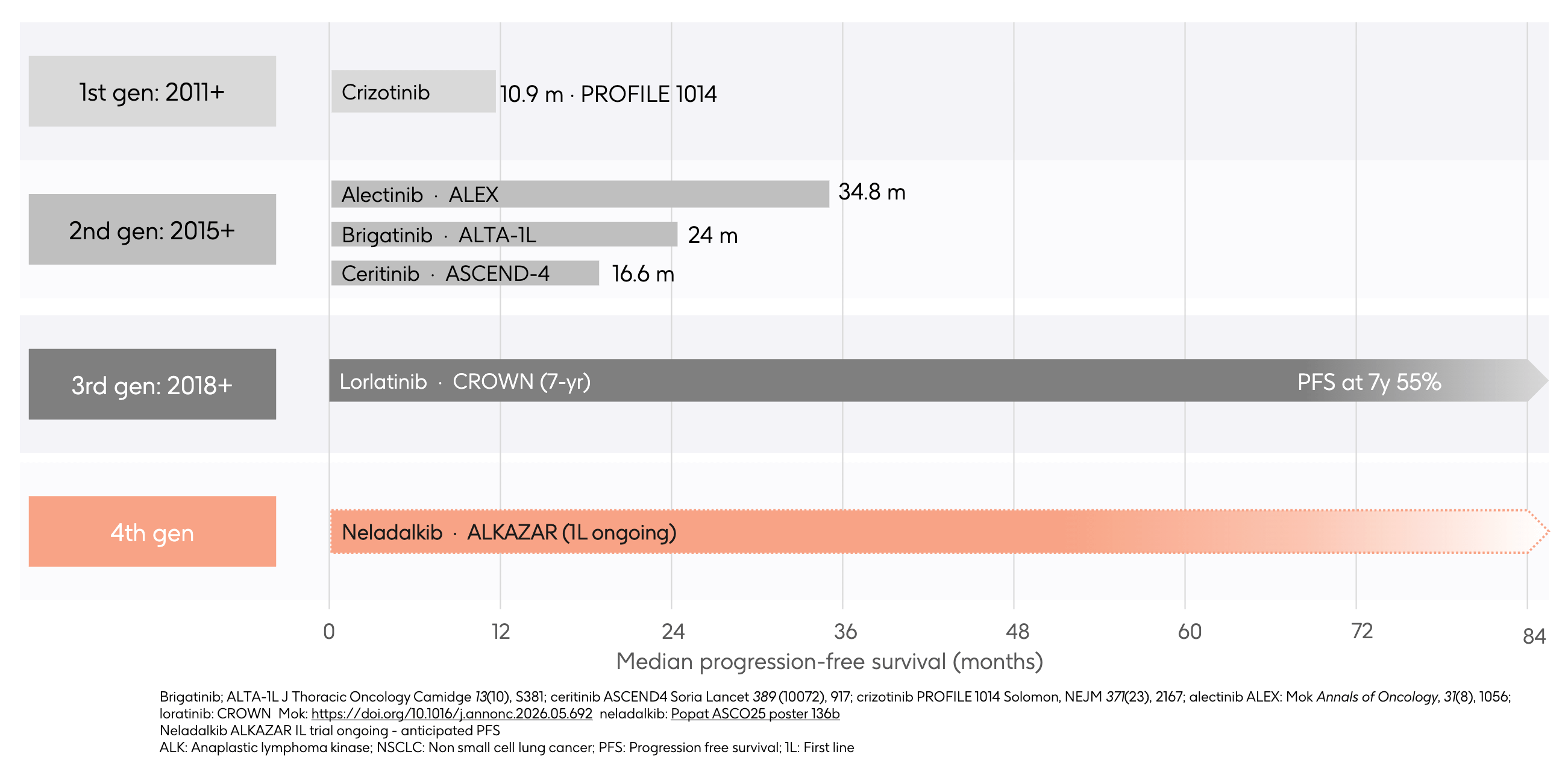
The ALK story began in earnest when crizotinib, originally developed as a MET inhibitor, was discovered to have potent activity against the EML4-ALK fusion oncogene. It cleared the regulatory hurdle in 2011, providing an immediate, dramatic alternative to cytotoxic chemotherapy. While revolutionary, crizotinib had two flaws. First, it was a poor substrate for crossing the blood-brain barrier, leaving the central nervous system (CNS) as a wide-open sanctuary for brain metastases. Second, tumors rapidly evolved single, acquired point mutations in the ALK kinase domain (like the L1196M gatekeeper mutation), which physically blocked crizotinib from binding and triggered clinical relapse in under a year.
Second-generation ALK inhibitors were designed from the ground up to address crizotinib’s shortfalls. They featured altered chemical scaffolds that achieved significantly greater potency against primary resistance mutations and, crucially, were engineered to optimize central nervous system (CNS) penetrance. Alectinib eventually emerged as the undisputed king of this generation. As shown on the chart, the landmark ALEX trial demonstrated an unprecedented 34.8 months of mPFS, effectively delaying tumor progression for nearly three years in the front-line setting while aggressively preventing or shrinking brain metastases. Despite this massive step forward, tumors eventually outsmarted second-generation TKIs by developing a highly aggressive, bulky “solvent-front” mutation known as ALK G1202R. This mutation structurally altered the binding pocket, causing steric clash that blocked alectinib, brigatinib, and ceritinib entirely.
To crack the G1202R mutation, Pfizer engineered lorlatinib, a highly compact, macrocyclic 3rd-generation ALK inhibitor designed to fit deeply into the altered ALK binding pocket regardless of standard solvent-front mutations. The updated 7-year data from the CROWN trial represents an absolute summit in solid-tumor oncology: an astonishing 55% of patients remain progression-free at the 7-year mark in the first-line setting. However, lorlatinib’s structural triumph created a massive, distinct double-gap:
Compound Mutations: When tumors finally breakthrough lorlatinib, they do so by stacking mutations atop one another, developing complex compound mutations (such as G1202R combined with L1196M or I1171T) that no approved drug can inhibit.
Off-Target TRK Inhibition: Lorlatinib’s small, rigid structure lacks macro-selectivity. It inadvertently binds heavily to the TRK receptor family in the brain. This off-target binding causes severe, treatment-limiting neurological toxicities, including profound cognitive impairment, memory loss, severe mood fluctuations, and massive, rapid weight gain.
This brings the narrative to the modern era and GSK’s recent strategic acquisition of Nuvalent’s portfolio. Neladalkib (formerly NVL-655) represents the fourth generation of ALK inhibition, engineered specifically to pick up where lorlatinib’s limitations begin. Neladalkib utilizes a novel macrocyclic scaffold that is deliberately optimized to maintain extreme potency against the entire spectrum of single and compound resistance mutations (including the dreaded lorlatinib-resistant lines). Crucially, its structure is TRK-sparing, allowing it to achieve deep brain penetrance to treat intracranial disease without triggering the debilitating cognitive and neurological side effects that limit lorlatinib’s utility. With the global Phase 3 ALKAZAR trial actively evaluating neladalkib head-to-head against alectinib in the front-line setting, the timeline projects a future where the red shaded bar intends to redefine the ceiling of long-term survival by offering a highly differentiated, TRK-sparing safety profile and potent coverage against known resistance mutations for ALK-positive patients.
Bringing ROS1 into Focus
Unlike many oncogenes discovered directly in human tissue, ROS1 was first identified via veterinary virology. In 1981, researchers isolating the transforming component of the UR2 avian sarcoma virus, a retrovirus that caused tumors in chickens, discovered a unique viral oncogene sequence they named v-ros. By 1986, molecular biologists cloned the human cellular homolog, identifying the normal proto-oncogene on chromosome 6 (6q22.1) and labeling it c-ros (now standardized as ROS1). Phylogenetic analysis revealed that ROS1 encodes a massive, type I transmembrane receptor tyrosine kinase belonging to the insulin receptor superfamily. It is evolutionarily linked to the sevenless (sev) gene in Drosophila.
Despite its identification, ROS1 was designated an orphan receptor, a receptor with an unidentified endogenous ligand. Even today, no physiological, native ligand has been definitively established for human ROS1, making wild-type functional research notoriously difficult. Knockout mice models showed that missing the receptor entirely resulted in perfectly healthy, viable animals, save for localized epithelial defects causing male infertility.
The oncogenic capability of ROS1 in humans was first identified in 1987. Researchers trying to identify transforming DNA from human tumor lines transfection-isolated a unique chromosomal deletion in the U118MG glioblastoma (brain cancer) cell line. This microdeletion on chromosome 6 fused a gene called FIG (now standardized as GOPC) to the kinase domain of ROS1. The resulting GOPC-ROS1 fusion protein was localized to the Golgi apparatus, where it drove downstream signaling. However, much like the early ALK discoveries in lymphoma, glioblastoma fusions were deemed a rare, niche phenomenon, and ROS1 receded from the therapeutic spotlight for two decades.
A defining moment for ROS1 in solid tumors arrived in 2007, coincidentally the exact same year the EML4-ALK fusion was uncovered in lung cancer. Instead of focused gene sequencing, a team of global scientists utilized a cutting-edge, high-throughput phosphoproteomics screen to map out global tyrosine kinase signaling across dozens of non-small cell lung cancer (NSCLC) cell lines and patient tumors. They were looking for cells completely saturated with abnormal survival signaling. In a distinct subset of lung adenocarcinoma samples that tested completely negative for known drivers like EGFR, KRAS, or ALK, the screen detected massive, aberrant phosphorylation clustering directly on the ROS1 kinase domain. Genetic sequencing of these samples revealed brand-new, somatic chromosomal translocations that fused the 3’ region of the ROS1 gene (encoding the intact intracellular tyrosine kinase domain) to the 5’ regions of completely unrelated driver partners.
The most prominent of these newly discovered lung cancer fusions was CD74-ROS1, alongside others like SLC34A2-ROS1. The structural architecture of these fusions explains their ruthless oncogenic drive. The fusion partner (e.g., CD74) provides a structured domain, typically a coiled-coil or WD40 repeat domain, that naturally forces proteins to cluster together (multimerize). This forces the attached, intracellular ROS1 kinase domains into a state of perpetual, ligand-independent autophosphorylation. Locked permanently in the “on” position, the mutant ROS1 engine floods the cell with continuous, hyperactive signaling through the STAT3, PI3K/AKT/mTOR, and RAS/RAF/MAPK pathways, bypassing cellular checkpoints to drive rapid proliferation.
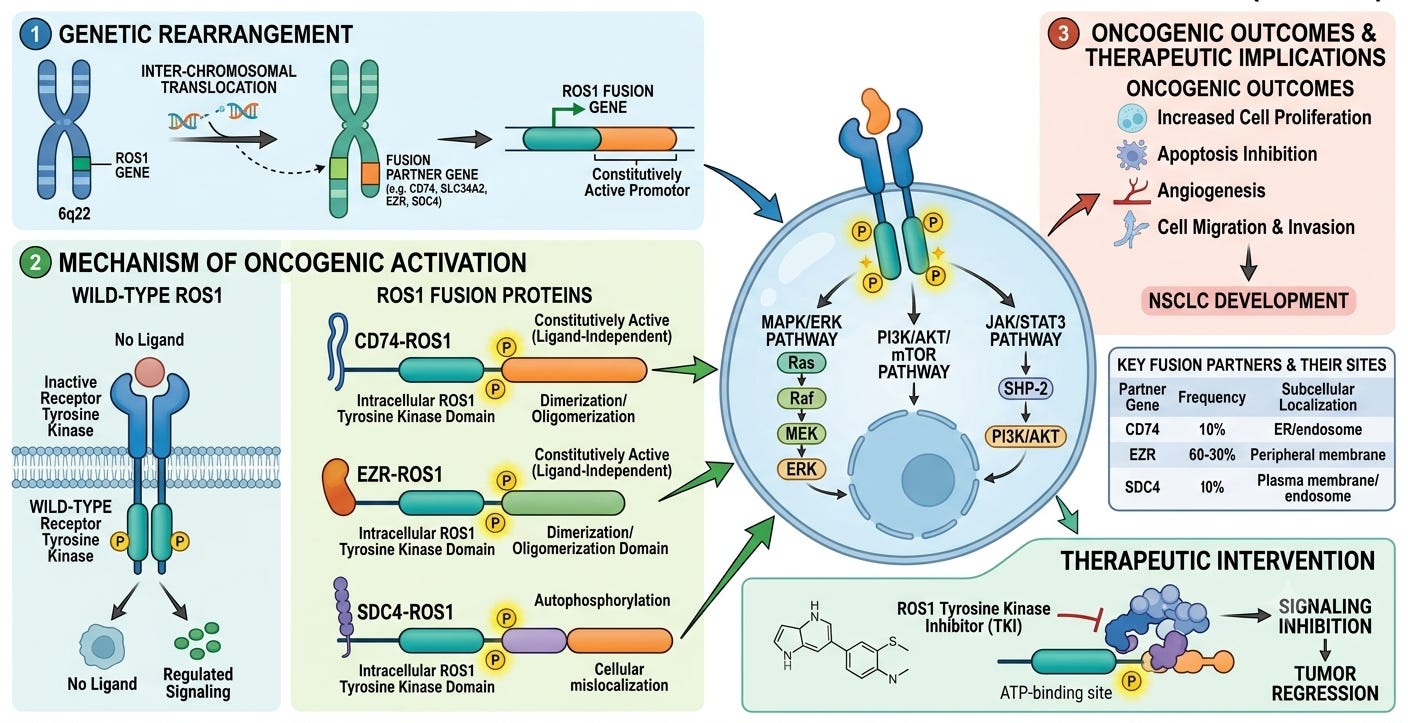
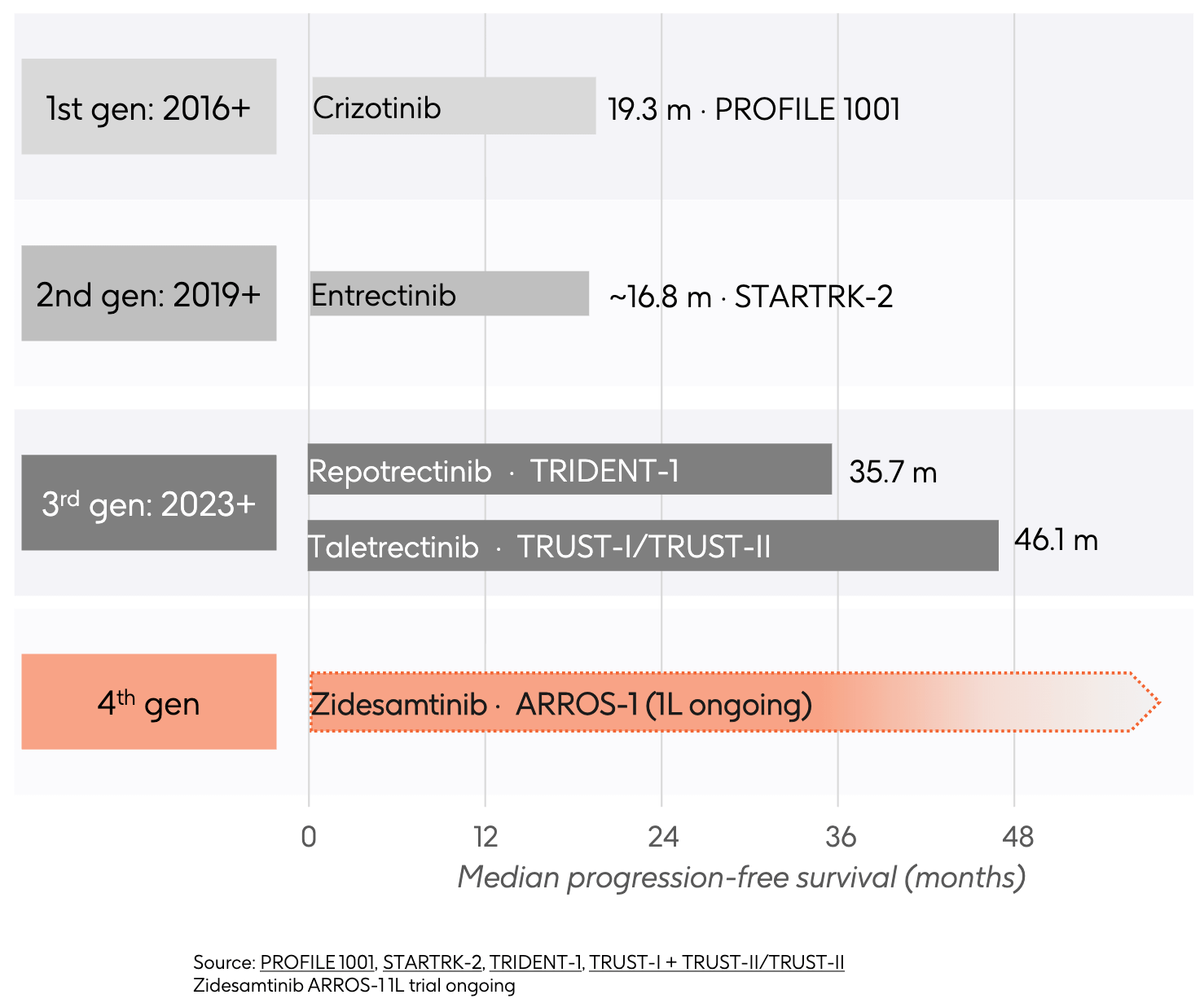
As large-scale epidemiological screening expanded, the ROS1 archetype crystallized. Comprising roughly 1% to 2% of all NSCLC cases, ROS1-positive patients matched the ALK clinical profile almost perfectly: they were typically younger (median age around 50), disproportionately female, and had little to no smoking history. Since the ATP-binding pocket of the ROS1 kinase domain shares massive amino acid homology with ALK, clinicians realized that small-molecule inhibitors designed to fit into the ALK pocket could likely cross-react and shut down ROS1. This biochemical crossover allowed crizotinib, already FDA-approved for ALK in 2011, to be rapidly deployed in clinical trials for ROS1 patients. By 2016, crizotinib secured expanded approval as the first targeted therapy for ROS1-positive NSCLC, setting off the targeted therapeutic arms race.
Much like its story in ALK, crizotinib fell short on two fronts. It suffered from poor blood-brain barrier penetrance, allowing brain metastases to emerge or progress unchecked. Furthermore, tumors rapidly escaped crizotinib by developing the ROS1 G2032R solvent-front mutation. This mutation introduces a bulky arginine residue that physically blocks crizotinib from entering the binding pocket. Approved in 2019, second-generation ROS1 inhibitor entrectinib was intentionally designed to be a potent, CNS-active inhibitor of both ROS1 and TRK. In the STARTRK-2 trial, it established strong efficacy against baseline brain metastases, addressing crizotinib’s delivery flaw. Despite its superior brain penetrance, entrectinib’s overall first-line mPFS (~16.8 months) didn’t eclipse crizotinib due to early resistance. More critically, entrectinib remained entirely inactive against the emergent G2032R solvent-front mutation. Additionally, because it explicitly targets the TRK receptor family, patients frequently suffered from off-target neurological toxicities like severe dizziness, cognitive changes, and paresthesia.
To tackle the crizotinib- and entrectinib-resistant tumors, the third generation ROS1 inhibitors introduced highly rigid, smaller macrocyclic or rigid chemical structures designed to squeeze past the bulky G2032R mutation. This generation raised the ROS1 efficacy ceiling. Repotrectinib delivered an impressive 35.7 months mPFS, while taletrectinib pushed the frontier even further to an astonishing 46.1 months mPFS in TKI-naive patients. Both drugs offered potent, dual-pronged protection: they effectively shut down the primary G2032R resistance mutation and demonstrated exceptional intracranial activity. Even with this elite efficacy, a double-gap remained:
Next-Gen Resistance: Under the selective pressure of 3rd-generation TKIs, tumors inevitably adapt by developing novel, complex compound mutations or specific “gatekeeper” and “solvent-front” doublets (such as L2026M or G2032R combinations) that these drugs fail to inhibit.
TRK Co-Inhibition: Repotrectinib is a highly potent pan-TRK inhibitor. Its rigid, compact structure binds tightly to TRK receptors in the brain, making side effects like profound dizziness (ataxia), dysgeusia (taste distortion), and paresthesia a significant, chronic management challenge for patients.
This culminates in the modern frontier and GSK’s strategic acquisition of Nuvalent’s portfolio. Zidesamtinib (formerly NVL-520) was engineered from the ground up as a 4th-generation ROS1 inhibitor, a TRK-sparing macrocycle. Nuvalent designed zidesamtinib’s structure to fit cleanly into both wild-type ROS1 and the mutated G2032R pocket, but gave it a precise conformation that causes structural clash with TRK receptors. This is intended to allow zidesamtinib to maintain high potency against specific resistance mutations and exhibit deep brain penetrance with reduced TRK-mediated neurological side effects relative to earlier-generation therapies. With its PDUFA date fast approaching on September 18, 2026, and the first-line ARROS-1 trial underway, the timeline represents a future where the red shaded bar aims to redefine long-term, toxicity-free survival for ROS1-positive patients.
Making Resistance Futile
The story of Nuvalent is a story of precision chemistry, born out of a desire to overcome drug resistance. James Porter, an exceptional medicinal chemist, and Alexander Fraley founded Nuvalent with a deceptively simple observation: early-generation tyrosine kinase inhibitors (TKIs) were leaving too many patients behind. In the rush to develop targeted therapies for oncogene-driven cancers, like ALK-positive and ROS1-positive non-small cell lung cancer (NSCLC), the industry had settled for structural compromises. Backed by the company creation experts at Deerfield Management, Nuvalent set to crafting rigid, low-molecular-weight molecules from the ground up.
By 2021, Nuvalent was ready to show its hand. Under the leadership of Chief Executive Officer Dr. Christopher Turner, the company went public in a highly successful $174 million IPO. The capital allowed them to transition from a fascinating chemistry hypothesis into a clinical-stage reality. Their twin lead candidates, NVL-520 (later named zidesamtinib) for ROS1 and NVL-655 (later named neladalkib) for ALK, entered the clinic. Over the next three years, data from their ARROS-1 and ALKAY can clinical trials began trickling out at major medical conferences like ASCO and ESMO. The oncology community watched with mounting excitement as the data proved Porter’s thesis correct. In reported clinical cohorts, these candidates demonstrated encouraging objective response rates, including intracranial activity, accompanied by a highly differentiated safety profile characterized by low rates of TRK-associated neurological adverse events. Nuvalent had successfully built TRK-sparing inhibitors that could outsmart the most notorious resistance mutations, like ROS1 G2032R and ALK G1202R. By early 2024, the FDA recognized the profound potential of this platform, granting Breakthrough Therapy designations to both lead assets. Nuvalent transformed into a formidable oncology contender racing toward the regulatory finish line.
As 2025 turned into 2026, Nuvalent entered its most intense operational phase. The company advanced a third asset, NVL-330, into Phase 1 clinical trials to target HER2-mutant lung cancers, proving that their design philosophy could be replicated across multiple distinct oncogenes. Zidesamtinib’s New Drug Application (NDA) was accepted by the FDA with a critical PDUFA action date set for September 18, 2026, and neladalkib was swiftly moving through a global Phase 3 trial (ALKAZAR) while its own priority review clock ticked toward a November 2026 deadline. Nuvalent had reached the ultimate crossroads for a successful independent biotech: it possessed a validated, multi-asset pipeline, but lacked the global commercial muscle required to launch three oncology drugs simultaneously.
The climax of the Nuvalent story arrived in June 2026. Recognizing the rare brilliance of a pipeline that had systematically “solved” the biggest efficacy and tolerability gaps in thoracic oncology, pharmaceutical giant GSK stepped forward. In a definitive $10.6 billion acquisition, GSK absorbed Nuvalent, framing the transaction to their investors as the ultimate “multiple assets in one deal” strategic victory. What began eight years prior as a chemist’s conviction in a Boston laboratory concluded as one of the most successful biotechnology narratives of the decade, handing GSK a ready-made, powerhouse lung cancer franchise and cementing Nuvalent’s legacy as the team that redefined the limits of targeted precision medicine.
Conclusion
The $10.6 billion acquisition of Nuvalent stands as an informative case study, demonstrating that the ultimate value in drug development often lies not in discovering a brand-new target, but in mastering the molecular architecture required to fix its flaws. By securing a powerhouse trio of potentially best-in-class assets in zidesamtinib, neladalkib, and NVL-330, GSK circumvents foundational biological uncertainty, establishes immediate commercial scale, and seamlessly anchors its broader thoracic oncology footprint. As the regulatory clocks tick closer to the late-2026 PDUFA dates for both zidesamtinib and neladalkib, the industry will be watching closely. What began as a chemist’s conviction in a Boston laboratory has officially evolved into a premier lung cancer franchise, one that is poised to redefine the upper limits of long-term, toxicity-free survival for patients worldwide.
To contact us, please send us an email at biotechreadout@gmail.com
Disclaimers
Investigational Status Disclaimer
The therapeutic candidates discussed in this newsletter are currently in clinical development and have not been approved for commercial sale by the U.S. Food and Drug Administration (FDA), the European Medicines Agency (EMA), or other global regulatory authorities. Their safety and efficacy have not been established. References to pipeline products and ongoing clinical trials involve significant risks and uncertainties. Statements regarding the potential safety, potency, or efficacy of investigational drugs reflect current hypotheses and are not a guarantee of future performance or regulatory clearance. The outcome of clinical trials is inherently unpredictable, and clinical results from earlier stages may not be predictive of results in later, larger-scale trials.
No Medical Advice Disclaimer
This newsletter is for informational and educational purposes only. The content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or another qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read in this publication.
No Patient-Provider Relationship Disclaimer
The information provided in this newsletter is for educational and analytical purposes only. Receipt of this information, or any interaction with this content, does not create a physician-patient, pharmacist-patient, or any other professional-provider relationship between you and the authors or publishers. This newsletter should not be used as a substitute for a personal consultation with a qualified healthcare professional.
Forward-Looking Statements Disclaimer
This newsletter contains “forward-looking statements” regarding future events, including clinical trial timing, regulatory milestones, and projected market performance. These statements are based on current expectations and assumptions that are subject to significant risks and uncertainties. Actual results may differ materially from those expressed or implied. We undertake no obligation to update these statements as a result of new information or future developments.
Third-Party Links & Content Disclaimer
This newsletter contains links to third-party websites, including clinical trial registries and corporate presentations. Biotech Readout does not endorse, guarantee, or assume responsibility for the accuracy or reliability of any information offered by third-party providers.
Errors and Omissions Disclaimer
While we strive for technical accuracy, the information in this newsletter is provided on an “as is” basis with no guarantees of completeness, accuracy, or timeliness. Biotech Readout assumes no liability for any errors or omissions in the content of this publication.
Non-Endorsement Disclaimer
Any reference to specific commercial products, processes, or services by trade name, trademark, or manufacturer does not constitute or imply an endorsement or recommendation by the author. All trademarks are the property of their respective owners.
No Investment Advice Disclaimer
This newsletter is for informational purposes only and does not constitute financial, investment, or legal advice. The author is not a registered investment advisor. You should consult with a professional financial advisor before making any investment decisions. The biotechnology sector is highly volatile; past performance is not indicative of future results.
Conflict of Interest Disclaimer
The author of this newsletter maintains a position of independence. At the time of publication, the author holds no direct financial interest, equity, or options in any of the companies mentioned in this report. No compensation has been received from any third party to feature or analyze specific therapeutic candidates or corporate entities.