
Chiesi to Acquire KalVista
An oral HAE play

Disclaimer: This newsletter is for educational and informational purposes only and does not constitute medical, investment, or financial advice, nor does it establish a provider-patient relationship. Content may include forward-looking statements and discussions of investigational therapeutic candidates that are not FDA/EMA approved; their safety and efficacy remain unestablished and clinical outcomes are unpredictable. While we strive for accuracy, all information is provided as is without guarantees. This newsletter is independent, and the author holds no financial positions in the companies mentioned nor receives third-party compensation for this coverage. Revenue run rates are estimates based on the author’s synthesis of publicly available 10-K/10-Q filings and may not reflect GAAP-certified annual totals. Please find a complete version of our disclaimers at the bottom of this article and on our About page.

Introduction
On April 29th, 2026, KalVista announced a definitive agreement that Chiesi Group would acquire them for approximately $1.9 billion, representing a 36% premium over the 30-day volume-weighted average share price. The centerpiece of this acquisition is Ekterly (plasma kallikrein inhibitor), which is the first FDA-approved oral, on-demand treatment for hereditary angioedema (HAE), a rare condition characterized by dangerous swelling. Clinical data from the Phase 3 KONFIDENT trial published in NEJM met its primary endpoint, demonstrating a statistically significant reduction in time to symptom relief compared to placebo in the trial population. By acquiring KalVista, Chiesi aims to expand its rare disease portfolio and significantly grow its commercial presence in the United States. Additionally, medical guidelines (2026 International Guideline on the Diagnosis and Management of Pediatric Patients with Hereditary Angioedema) were updated by the issuing medical committee to include Ekterly as a ‘strong recommendation’ for first-line therapy for the acute treatment of HAE attacks in adolescents aged 12 years and older. This transition from injectable treatments to an oral option is described as a major advancement for patient independence and early intervention.
HAE constitutes a blockbuster indication space, representing an aggregate revenue run rate of almost $2.5 billion according to quarterly press releases for 4Q 2025 associated with Takhyzro, Orladeyo, Ekterly, Firazyr, Cinryze*.
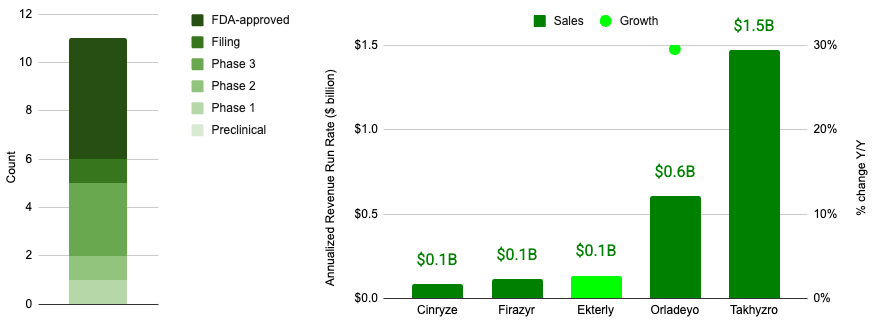
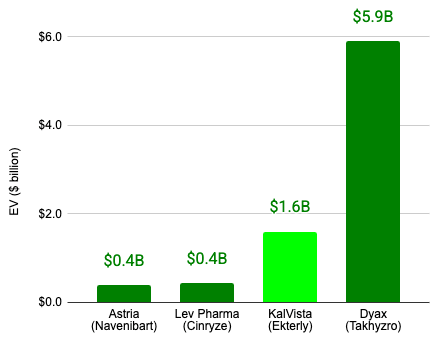
If executed, the proposed KalVista acquisition would constitute the second largest acquisition in the HAE space to date.
Here, I dive into the history of hereditary angioedema (HAE), the evolution of therapeutic approaches, and the story of KalVista.
What is HAE?
Not everyone is acquainted with HAE (after all, it is a rare disease), so allow me to provide a short explainer. Hereditary Angioedema (HAE) is a rare genetic disorder characterized by recurrent episodes of severe swelling (angioedema) that impacts approximately 10,000 people in the United States. Unlike common allergic reactions, HAE does not respond to antihistamines or epinephrine because it is driven by an overproduction of bradykinin, a peptide that causes blood vessels to leak fluid into surrounding tissues. HAE attacks typically manifest as non-pitting edema, meaning if you press a finger into the swollen area, it doesn’t leave a lasting indentation. Attacks impacts a number of body parts:
Skin: The skin often looks pale or slightly pink, but it is not usually accompanied by hives (urticaria). If you see itchy, red welts, it is likely an allergy rather than HAE. It most frequently affects the hands, feet, face, and genitals. A hand can swell to double its size, making it impossible to grip objects or wear rings. Some patients experience a prodrome before the swelling starts, most notably Erythema Marginatum, a non-itchy, map-like red rash that looks like marble patterns on the skin.
Gastrointestinal Tract: While you can’t see this from the outside, the lining of the intestines swells significantly. This causes the abdomen to look distended or bloated. The pain is often described as excruciating and is frequently mistaken for appendicitis or a ruptured organ. It is often accompanied by severe nausea and vomiting.
Upper Airway: This appears as swelling of the tongue, soft palate, and throat. This is the most life-threatening form of HAE. It can progress quickly, leading to a tight feeling in the throat, difficulty swallowing, or a change in voice (hoarseness), eventually risking total airway obstruction.
HAE is also classified into three types:
Type I (80-85% of cases): This is the most common type, in which the body doesn’t produce enough C1-Inhibitor protein.
Type II (15-20% of cases): The body produces enough C1-Inhibitor, but the protein doesn’t function correctly.
HAE with Normal C1-INH (<1% of cases): A rarer form where the C1 protein is fine, but other genetic mutations (like Factor XII) cause the same bradykinin buildup.
Despite its prevalence, the underdiagnosed nature of the disease causes a significant lag in these numbers. On average, it still takes 8 to 10 years for a patient to receive an accurate HAE diagnosis after their first attack. Approximately 25% of cases are the result of a spontaneous de novo mutation, meaning there is no family history to help guide the diagnosis.
A Swelling Suspicion
The history of Hereditary Angioedema (HAE) is a fascinating journey from early clinical observations to a sophisticated understanding of molecular genetics and targeted therapies. While early accounts of swelling go back to the 16th century, the disease was first formally distinguished from standard hives in the late 19th century. In 1882, Dr. Heinrich Quincke first described the clinical symptoms of angioedema, often referred to at the time as Quincke’s edema. A few years later in 1888, Dr. William Osler provided the definitive description of the hereditary nature of the disease. He tracked the condition through five generations of a family and famously named it Hereditary Angioneurotic Edema (HANE), a name that stuck for decades until the neurotic component was debunked.
For 75 years following Dr. Osler’s report, the underlying cause of HAE remained a mystery, and mortality rates from laryngeal edema were as high as 25–30%. Then in 1963, Virginia Donaldson and Jack Evans at Case Western Reserve University identified that HAE is caused by a deficiency in the C1 esterase inhibitor (C1-INH). This established HAE as the first human disease recognized to be caused by a deficiency of a complement control protein.
Before their work, scientists knew about the C1 esterase inhibitor (C1-INH), a protein that keeps the body’s complement system (part of the immune response) in check. However, no one had linked a deficiency of this protein to a specific human disease. The year before Donaldson’s breakthrough, a researcher named Nathaniel Landerman working at the University of Maryland School of Medicine published a study (1962) showing that patients with HAE were missing a specific “inhibitor of permeability” in their blood. Landerman found that these patients couldn’t properly shut down kallikrein, an enzyme that causes blood vessels to leak. This proved that HAE was a disease of missing brakes, a lack of a regulatory protein rather than the presence of a toxin.
While Landerman was looking at HAE, a scientist named Irwin Lepow at Case Western Reserve University had just discovered and characterized a protein that inhibited the first component of the complement system (C1). He called it C1-esterase inhibitor. Since Donaldson was working at the same institution and in the same scientific circles as Lepow, she was aware of this newly identified inhibitor protein. She and Evans theorized that the kallikrein inhibitor Landerman had described might actually be the same protein Lepow had just discovered.
Donaldson and Evans tested the serum of several patients with HAE. Indeed, they discovered that these patients completely lacked the serum globulin that inhibits the enzymatic activity of C1 esterase. Interestingly, Donaldson and Evans initially believed the swelling was caused by the complement system (since they found a C1 inhibitor deficiency), specifically a downstream fragment called C2 kinin. Research eventually showed that C2 kinin was too weak to cause the massive, localized edema seen in HAE patients. Furthermore, antihistamines and steroids (which usually work on immune-driven swelling) were useless, suggesting the immune explanation was incomplete.
In the late 1970s and early 80s, researchers began to look more closely at the Contact System (also known as the Kallikrein-Kinin system), which governs blood pressure and vascular permeability. Researchers realized that C1-INH was not just an inhibitor of C1. It was actually a broad-spectrum serine protease inhibitor (serpin) that regulated multiple enzymes, including Plasma Kallikrein and Factor XII. It was discovered that when C1-INH is missing, Plasma Kallikrein goes into overdrive. This enzyme cleaves a large protein called High-Molecular-Weight Kininogen (HMWK), and the byproduct of that cleavage is Bradykinin.
Bradykinin is a potent vasodilator, far more powerful than histamine. Its discovery as the primary mediator changed everything because of how it interacts with the blood vessels:
Binding the B2 Receptor: Bradykinin binds to B2 receptors on the endothelial cells lining the blood vessels.
Opening the Floodgates: Once bound, it signals the cells to pull apart slightly, creating gaps in the vessel wall. Plasma (the liquid part of the blood) leaks through these gaps into the surrounding tissue, causing the massive “non-pitting” swelling characteristic of HAE.
No Itch, Just Pain: Unlike histamine, bradykinin doesn’t stimulate the nerves that cause itching. Instead, it stimulates pain receptors and causes significant fluid shifts, explaining why HAE attacks are painful rather than itchy.
Today, we have a more complete picture of the mechanism that cause HAE. It is primarily driven by a deficiency or dysfunction of the C1 esterase inhibitor (C1-INH), leading to chronically overactive plasma kallikrein. This triggers uncontrolled cleavage of high-molecular-weight kininogen and sets off the excessive release of bradykinin, a potent inflammatory mediator that binds to B2 receptors on vascular endothelial cells. This binding signals the endothelial cells to retract, increasing vascular permeability and allowing plasma to leak into the surrounding interstitial tissues, which manifests clinically as the localized, non-pitting, and potentially life-threatening swelling characteristic of the disease.
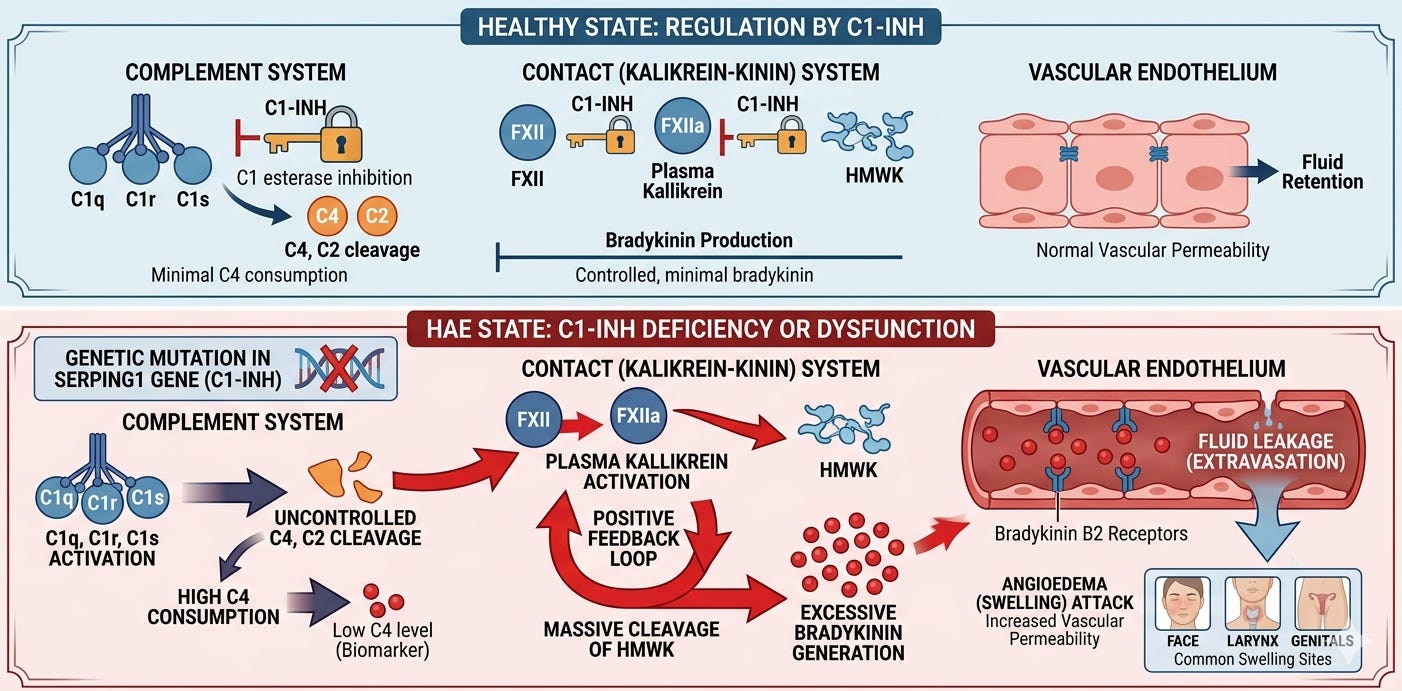
This shift in understanding allowed the industry to move away from non-specific therapies and toward precise molecular targets.
From Blunt Instruments to Precision Strikes
The history of HAE therapy is a transition from brute force medicine, using repurposed steroids and blood products, to highly elegant, targeted molecular engineering. In the 1950s-1970s, before the molecular cause of HAE was understood, doctors relied on trial and error with medications designed for other conditions:
Antifibrinolytics (Epsilon-aminocaproic acid & Tranexamic acid): Originally used to control bleeding, these were found to modestly reduce HAE attacks. They worked by indirectly slowing down the activation of the contact system, but they were often insufficient for severe cases.
Fresh Frozen Plasma (FFP): In emergencies, doctors administered FFP to provide a top-off of C1-inhibitor. However, this was a double-edged sword: FFP also contains the substrates (like kininogen) that produce bradykinin, occasionally causing a paradoxical worsening of the attack before it got better.
Methyltestosterone: Early observations showed that high-dose male hormones could reduce swelling, though the side effects were often as debilitating as the disease itself.
Following the 1963 discovery of the C1-INH deficiency, researchers looked for ways to turn on the body’s own production of the protein. In this vein, attenuated androgens (Danocrine, Stanozolol) were approved in the mid-1970s. These became the first-line long-term prophylaxis for decades. They stimulate the liver to increase the synthesis of C1-INH. While effective, they caused significant long-term side effects: weight gain, acne, virilization in women (deepening voice, hair growth), and a long-term risk of liver tumors (adenomas).
As biotechnology advanced, the focus shifted from stimulating the liver to simply replacing the missing protein directly. Plasma-derived C1-INH were extracted and highly purified from human donor plasma. In 2008, Cinryze became the first FDA-approved drug for preventing HAE attacks (prophylaxis). One year later in 2009, Berinert was approved for the acute treatment of attacks. To remove the reliance on human plasma, scientists engineered transgenic rabbits to produce human C1-INH in their milk, providing a highly concentrated alternative. In 2014, Ruconest became the first and only recombinant (plasma-free) C1-INH treatment available in the United States.
Once the Bradykinin Hypothesis was solidified, therapies began targeting the specific molecules that cause the leak:
B2 Receptor Antagonists (Firazyr approved in 2009): Instead of stopping the production of bradykinin, this drug acts as a shield on the blood vessels, preventing bradykinin from docking and opening the floodgates.
Plasma Kallikrein Inhibitors (Kalbitor approved in 2011): A potent inhibitor that stops the enzyme responsible for generating bradykinin in the first place.
As therapies improved, the goal shifted from surviving attacks to total freedom from attacks using long-acting biologics:
Monoclonal Antibodies (Takhzyro approved in 2018): By specifically binding to and inhibiting active plasma kallikrein with a long half-life, it allowed patients to go from daily or weekly treatments to a single injection every two or four weeks.
Oral Small Molecules (Orladeyo approved in 2020): This provided the first non-steroidal daily pill to prevent attacks, significantly reducing the treatment burden of needles and infusions.
KalVista, a Successful Pivot
The history of KalVista Pharmaceuticals is a study in strategic pivot and clinical persistence, culminating in the first oral on-demand treatment for HAE. While the company originally explored ophthalmology, its refocus on the kallikrein-kinin system eventually led to its acquisition by the Chiesi Group on April 29, 2026.
KalVista was formally launched in 2011 (though its U.S. entity was incorporated in 2004) by T. Andrew Crockett and experts from Harvard Medical School. The company began as an ophthalmology-focused entity, targeting Diabetic Macular Edema (DME). The team focused on small-molecule protease inhibitors. Their early lead candidate, KVD001, was an intravitreal injection designed to block plasma kallikrein in the eye. In November 2016, KalVista went public on the NASDAQ via a reverse merger with Carbylan Therapeutics.
As the company advanced its protease platform, it became clear that their oral kallikrein inhibitors had massive potential for systemic diseases like Hereditary Angioedema. They began developing KVD900 (now sebetralstat) for acute attacks and KVD824 for prophylaxis. In 2019, the DME program (KVD001) failed its Phase 2 trial. Shortly after, the prophylaxis candidate (KVD824) faced a temporary FDA clinical hold and was eventually terminated in 2022 due to liver enzyme elevations. Following these setbacks, KalVista pivoted entirely to HAE, focusing all resources on sebetralstat (an oral plasma kallikrein inhibitor) as a potential paradigm shifter for on-demand care.
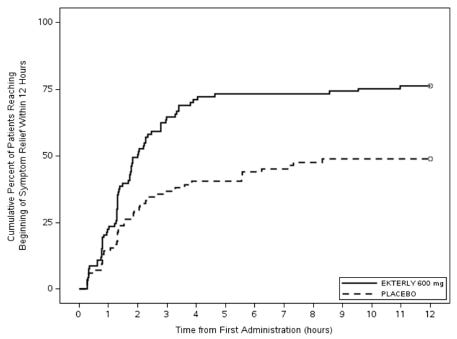
The strategy to focus on oral on-demand treatment proved successful, addressing a major unmet need for patients who wanted to avoid injections during an attack. In February 2024, KalVista announced positive topline results from the pivotal Phase 3 KONFIDENT trial. The data showed that sebetralstat significantly reduced time to symptom relief compared to placebo (p < 0.001). The primary endpoint for KONFIDENT was the ‘time to beginning of symptom relief’ defined as at least “a little better” at two consecutive time points within 12 hours of first dose administration, assessed using a seven-point scale Patient Reported Global Impression of Change (PGI-C) ranging from “much worse” to “much better”. A total of 71 out of 93 (76%) patients administered sebetralstat 600 mg and 41 out of 84 (49%) patients administered placebo achieved the primary endpoint. The median time to beginning of symptom relief within 12 hours of first dose was 2.0 hours (95% CI: 1.5, 2.8) in patients administered sebetralstat 600 mg.
On July 3, 2025, the FDA approved Ekterly (sebetralstat) for the on-demand treatment of HAE in patients aged 12 and older. By late 2025 and early 2026, the drug received approvals in the EU, UK, Switzerland, and Japan, establishing KalVista as a global commercial-stage rare disease company.
On March 25, 2026, KalVista reported its financial results for the eight-month transition period ending December 31, 2025. The report highlights the successful commercial launch of Ekterly, the first oral on-demand treatment for HAE. The company generated $49.1 million in global net product revenue during the eight-month period following the U.S. launch on July 7, 2025. KalVista received a cumulative 1,702 patient start forms in the U.S. through February 2026, representing approximately 20% of the U.S. HAE patient population, according to the company.
KalVista also announced that they had completed enrollment for the open-label Phase 3 KONFIDENT-KID trial for children aged 2–11. In this demographic, the trauma of needles often leads to dangerous treatment delays. Currently, the only approved on-demand treatment for this age group in the U.S. is intravenous. Sebetralstat aims to be the first oral on-demand option, reducing the burden and anxiety associated with injections.
Less than a week later on March 30, 2026, KalVista reported interim findings in the KONFIDENT-KID trial (ages 2–11), which utilized a proprietary weight-based oral disintegrating tablet (ODT) formulation. As of December 15, 2025, data from 172 HAE attacks in 33 participants showed:
Rapid Treatment: The median time to treatment was 25 minutes, with 67% of attacks treated within the first hour.
Symptom Relief: In the largest cohort (150 mg dose), the median time to symptom relief was 1.5 hours, and complete resolution was achieved in 12 hours.
Safety: In this interim analysis of the KONFIDENT-KID trial, sebetralstat was well tolerated, with no reports of serious or treatment-related adverse events observed among participants to date and no difficulty swallowing the ODT.
KalVista expressed plans to submit a New Drug Application (NDA) in the U.S. in Q3 2026, with a company-projected launch in 2027, subject to FDA clearance. A little more than a month later on April 29th, 2026, KalVista and Chiesi Group announced their acquisition agreement. Chiesi Group’s acquisition of KalVista was primarily motivated by a strategic objective to expand its global rare disease portfolio. They viewed Ekterly as a “differentiated” therapy that provides a potentially foundational advancement in HAE management by enabling earlier intervention and reducing the overall treatment burden. The Chiesi team cited Ekterly as being a potential “a key driver in helping Chiesi reach its 2030 strategic revenue target of €6 billion”, suggesting that they were impressed with its launch performance. Lastly, they signaled its role in “expanding [their] commercial infrastructure and market presence in the United States”.
Conclusion
From Dr. Osler’s first observations in 1888 to the molecular discovery of the C1-inhibitor in a Cleveland lab, the story of HAE has always been one of “missing brakes.” KalVista’s journey, from its early pivots in ophthalmology to the successful launch of the first oral on-demand therapy, adds the latest chapter to that history. For many, the therapeutic landscape is shifting away from complex infusions toward the option of an oral tablet. It is a fitting conclusion to KalVista’s independent chapter and a promising hope for HAE patients.
To contact us, please send us an email at biotechreadout@gmail.com
Disclaimers
Investigational Status Disclaimer
The therapeutic candidates discussed in this newsletter are currently in clinical development and have not been approved for commercial sale by the U.S. Food and Drug Administration (FDA), the European Medicines Agency (EMA), or other global regulatory authorities. Their safety and efficacy have not been established. References to pipeline products and ongoing clinical trials involve significant risks and uncertainties. Statements regarding the potential safety, potency, or efficacy of investigational drugs reflect current hypotheses and are not a guarantee of future performance or regulatory clearance. The outcome of clinical trials is inherently unpredictable, and clinical results from earlier stages may not be predictive of results in later, larger-scale trials.
No Medical Advice Disclaimer
This newsletter is for informational and educational purposes only. The content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or another qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read in this publication.
No Patient-Provider Relationship Disclaimer
The information provided in this newsletter is for educational and analytical purposes only. Receipt of this information, or any interaction with this content, does not create a physician-patient, pharmacist-patient, or any other professional-provider relationship between you and the authors or publishers. This newsletter should not be used as a substitute for a personal consultation with a qualified healthcare professional.
Forward-Looking Statements Disclaimer
This newsletter contains “forward-looking statements” regarding future events, including clinical trial timing, regulatory milestones, and projected market performance. These statements are based on current expectations and assumptions that are subject to significant risks and uncertainties. Actual results may differ materially from those expressed or implied. We undertake no obligation to update these statements as a result of new information or future developments.
Third-Party Links & Content Disclaimer
This newsletter contains links to third-party websites, including clinical trial registries and corporate presentations. Biotech Readout does not endorse, guarantee, or assume responsibility for the accuracy or reliability of any information offered by third-party providers.
Errors and Omissions Disclaimer
While we strive for technical accuracy, the information in this newsletter is provided on an “as is” basis with no guarantees of completeness, accuracy, or timeliness. Biotech Readout assumes no liability for any errors or omissions in the content of this publication.
Non-Endorsement Disclaimer
Any reference to specific commercial products, processes, or services by trade name, trademark, or manufacturer does not constitute or imply an endorsement or recommendation by the author. All trademarks are the property of their respective owners.
No Investment Advice Disclaimer
This newsletter is for informational purposes only and does not constitute financial, investment, or legal advice. The author is not a registered investment advisor. You should consult with a professional financial advisor before making any investment decisions. The biotechnology sector is highly volatile; past performance is not indicative of future results.
Conflict of Interest Disclaimer
The author of this newsletter maintains a position of independence. At the time of publication, the author holds no direct financial interest, equity, or options in any of the companies mentioned in this report. No compensation has been received from any third party to feature or analyze specific therapeutic candidates or corporate entities.